بيلة الهيموجلوبين الليلية الانتيابية هي مرض نادر جدًا من مجموعة فقر الدم الانحلالي ولا يعتبر وراثيًا. ويتم اكتسابه خلال الحياة، على الرغم من أن له أساسًا وراثيًا. جوهر علم الأمراض هو التغيرات في بنية خلايا الدم (معظمها خلايا الدم الحمراء)، مما يؤدي إلى تدمير مبكر لأغشيتها وانهيار داخل الأوعية الدموية (انحلال الدم).
يبلغ معدل الانتشار حوالي 16 حالة لكل مليون نسمة، ويبلغ معدل الإصابة السنوي 1.3 لكل مليون في أغلب الأحيان، ولم يتم تحديد أي اعتماد على الجنس.
يتضمن الاسم أسماء الباحثين والأطباء الإيطاليين الذين أمضوا سنوات في الدراسة: مرض مارشيافافا-ميشيلي، مرض ستروبينج-مارشيافافا.
ما هي "بيلة الهيموجلوبين" وما أسبابها؟
بيلة الهيموجلوبين هي أحد أعراض أمراض مختلفة تسبب تكسير خلايا الدم الحمراء بسبب تأثيرها على الغشاء، بينما يترك الهيموجلوبين الخلايا ويدخل إلى البلازما.
في الشخص السليم، لا يمكن أن يكون أكثر من 5٪ من إجمالي حجم بلازما الدم. لوحظ زيادة في مستوى الهيموجلوبين بنسبة 20-25٪ في الاضطرابات الخلقية أو اعتلال الهيموجلوبين (ثلاسيميا بيتا، وتدمير الخلايا الحمراء في فقر الدم المنجلي).
تحدث بيلة الهيموجلوبين الشديدة بسبب الحالات التي يتم فيها تجاوز مستويات الهيموجلوبين المسموح بها بشكل كبير بسبب انحلال خلايا الدم الحمراء. نظام البلاعم غير قادر على معالجة مثل هذا الحجم الكبير من الصباغ، ويدخل الهيموجلوبين في البول.
يمكن أن تكون أسباب بيلة الهيموجلوبين:
- الأمراض المعدية الحادة (الأنفلونزا) ؛
- التهاب رئوي؛
- إصابات؛
- التسمم بسبب التسمم بأصباغ الأنيلين وحمض الكربوليك وملح البرثوليت.
- انخفاض حرارة الجسم الشديد.
- الإجهاد البدني القوي والمطول.
- نقل أنواع مختلفة من الدم.
- حروق واسعة النطاق
- تم تحديد دور الطفرة المكتسبة في جين PIG-A.
تستخدم أصباغ الأنيلين على نطاق واسع في صناعة النسيج وزخرفة الباتيك والتنظيف الجاف وخدمات الصباغة، ويتطلب العمل بها الحذر
لا توجد بيلة الهيموجلوبين دون ارتفاع مستوى الهيموجلوبين في الدم (هيموجلوبين الدم). ترتبط نوبات ما قبل الفجر بتحول فسيولوجي في التوازن الحمضي القاعدي نحو الحماض في الليل. يساهم المحتوى المتزايد من منتجات التحلل أيضًا في تحمض الجسم وزيادة انهيار خلايا الدم.
التسبب في الاضطرابات
تحدث التغيرات الرئيسية في بيلة الهيموجلوبين الليلية الانتيابية على مستوى المكمل. وهو يمثل سلسلة من التفاعلات الكيميائية الحيوية التي توفر مناعة فطرية.
تعتبر المادة الفعالة هي مجمع الهجوم الغشائي المشكل. يحتوي على حوالي 30 مكونًا منظمًا. يعتمد تركيب المكونات التكميلية على الإشارات الواردة من الجهاز العصبي والغدد الصماء. عادة، يتم التحكم فيه بواسطة بروتينات خاصة لا تسمح بتدمير الخلايا المضيفة (البشرية).
مع بيلة الهيموجلوبين الليلية، يتم فقدان هذه العملية. يتم تدمير الطبقة الدهنية من غشاء الخلية لخلايا الدم الحمراء، مما يسبب موتها. لقد ثبت زيادة حساسية غشاء كرات الدم الحمراء لتكملة المكونات.

المكمل ضروري لحماية الخلايا من العوامل المعدية والاستفادة من منتجات تحلل الكائنات الحية الدقيقة وخلاياها التالفة.
تتفاعل خلايا الدم الأخرى (كريات الدم البيضاء والصفائح الدموية) أيضًا عن طريق التسبب في حدوث عيوب في الغشاء. لم يتم العثور على تراكم الجلوبيولين المناعي عليها، مما يثبت عدم وجود آلية الحساسية الذاتية ويتحدث لصالح تلف الخلية السليفة المشتركة. هي التي تتلقى المعلومات الوراثية (الأمر) حول العمل المدمر.
تسمى المنطقة الجينية المفقودة للخلية الجذعية GPI-AP. يساهم نقصه في استنساخ كريات الدم الحمراء في التعرض لانحلال الدم تحت تأثير المكمل. وفي الوقت نفسه، يمكن أن يوجد استنساخ طبيعي لخلايا الدم الحمراء في الجسم.
تحدث بيلة الهيموجلوبين الليلية الانتيابية فقط إذا ساد الاستنساخ المرضي على الاستنساخ الطبيعي. يتم اكتشاف خلايا الدم الحمراء المأخوذة من نسخة مع غياب جزئي أو كامل لـ GPI-AP في المرضى عن طريق قياس التدفق الخلوي. من المهم ألا يكون عدد الخلايا المرضية لدى المرضى هو نفسه.
ترتبط زيادة تكوين الخثرة في مرض مارشيافافا-ميسيلي بتحفيز تخثر الدم عن طريق العوامل التي يتم إطلاقها أثناء تدمير خلايا الدم الحمراء.
أشكال المرض
يأخذ تصنيف الأشكال السريرية في الاعتبار البيانات المختبرية والعلاقة بين السبب والنتيجة لتغيرات الدم. من المعتاد التمييز بين الأصناف التالية:
- تحت الإكلينيكي - لا توجد علامات مختبرية لانحلال الدم، فقط الطرق الحساسة للغاية يمكنها اكتشاف عدد صغير من الخلايا التي تفتقر إلى GPI-AP. لا توجد صورة سريرية للمرض. غالبا ما يقترن بفقر الدم اللاتنسجي.
- الكلاسيكية - جميع الأعراض السريرية موجودة، وتحدث مع تفاقم دوري، بالإضافة إلى كريات الدم الحمراء، تتأثر كريات الدم البيضاء والصفائح الدموية، ويتم تحديد علامات انحلال الدم في المختبر (نمو الخلايا الشبكية، إنزيم هيدروجيناز اللاكتات في المصل، البيليروبين، مع انخفاض مستوى هابتوغلوبين). ولم يلاحظ أي تشوهات في تكون الدم في نخاع العظام.
- يحدث بسبب نقص تكوين الدم في نخاع العظم في أمراض مختلفة- يُفترض وجود أمراض مصاحبة أو سابقة لنخاع العظم مع اضطراب تكون الدم (مع فقر الدم اللاتنسجي، متلازمة خلل التنسج النقوي). يكشف التحليل والنتائج السريرية عن جميع مظاهر انحلال الدم على خلفية التشوهات في تكوين الدم في نخاع العظم.
ووفقا لتصنيف آخر يقترح التمييز بين:
- شكل مجهول السبب أو بيلة الهيموجلوبين الليلية الانتيابية نفسها ؛
- علم الأمراض في شكل متلازمة لمختلف الأمراض.
- وهو نوع نادرًا ما يتم ملاحظته ويحدث بعد نقص تنسج النخاع العظمي.
لا يوجد تصنيف يعتمد على مؤشر كمي لانتشار استنساخ غير طبيعي في الدم. لقد ثبت أن المسار تحت السريري ممكن مع استبدال الخلايا الطبيعية بنسبة 90٪. وفي مرضى آخرين، يحدث تجلط الدم الشديد في وجود 10٪ فقط من خلايا الدم الحمراء المتغيرة.
الأعراض والدورة السريرية
يمكن أن يبدأ المرض فجأة (بشكل حاد) أو يكون له مسار مزمن تدريجي. تسمى فترات التفاقم بالأزمات الانحلالية. غالبًا ما يسبقها نزلة برد سابقة أو ارتباط بالعدوى أو ملامسة مواد سامة.
تشمل الأعراض الرئيسية لبيلة الهيموجلوبين الليلية الانتيابية ما يلي:
- ألم المعدة؛
- ألم في الصدر بكثافة وتوطين متفاوتين - يرتبط الألم المتفاوت بالتوطين بتجلط الفروع الصغيرة للسرير الشرياني وتشكيل بؤر إقفارية في الأعضاء الداخلية.
- علامات فقر الدم (الضعف والدوخة والصداع) - الناجمة عن زيادة الدمار وعدم كفاية إنتاج خلايا الدم الحمراء، بالإضافة إلى أن الدراسات تشير إلى نقص الحديد وحمض الفوليك في دم المرضى؛
- اصفرار الجلد والصلبة - مؤشر على إطلاق البيليروبين المباشر في الدم، والذي يعالجه الكبد من الهيموجلوبين الزائد.
- اضطراب البلع.
- ضعف الانتصاب لدى الرجال - يتجلى ليس فقط على خلفية الأزمات، ولكنه يصبح مزمنا، بسبب انخفاض تركيز أكسيد النيتريك في البلازما، وضعف العضلات ونغمة الأوعية الدموية.
- زيادة التعب.
- ضيق في التنفس وخفقان.
- العلامات المحلية لالتهاب الوريد الخثاري (احمرار الجلد فوق الوريد، تورم، ألم عند الجس، زيادة في درجة الحرارة)؛
- عند فحص المريض، قد يلاحظ الطبيب تضخم الكبد والطحال، وهذه العلامة مهمة بشكل خاص لتشخيص تطور تجلط الدم والنوبات القلبية فيهما.
يساهم المسار المزمن للمرض في تطور:
- ارتفاع ضغط الدم الرئوي مع تجلط الدم في فروع الأوعية الرئوية.
- الفشل الكلوي المزمن الناجم عن ترسب منتج انهيار الهيموجلوبين (الهيموسيديرين) في الأنابيب الكلوية، تخثر الأوعية الدموية مع تشكيل احتشاءات مجهرية.
- حساسية عالية للعدوى المرتبطة بها.
تصبح هذه المتلازمات الأسباب الأكثر احتمالا للوفاة.
التشخيص المختبري
يتم تشخيص مرض مارشيافافا-ميشيلي بعد إجراء فحص شامل في مراكز أمراض الدم التي لديها القدرة على إجراء اختبارات وتحليلات محددة.
في الدم المحيطي تم العثور على:
- قلة الكريات الحمر، نقص الكريات البيض، نقص الصفيحات (حالة تثبيط النمو العام لخلايا الدم تسمى قلة الكريات الشاملة)؛
- كثرة الشبكيات.
- زيادة في مستوى الهيموجلوبين في البلازما.
- انخفاض مستويات الحديد والفولات.
يكشف فحص نخاع العظم عن:
- علامات تنشيط تكون الكريات الحمر (إنتاج خلايا الدم الحمراء) بسبب تراكم الخلايا السليفة (الخلايا الأرومية الطبيعية والبلازما والخلايا البدينة)؛
- انخفاض عدد الخلايا المحببة والخلايا المكروية.
- مناطق النزف، وتراكم خلايا الدم الحمراء المنحلّة في الجيوب الأنفية.
- في مرحلة قمع تكون الدم، تظهر مناطق الانحطاط الدهني والدمار.
اختبارات محددة تعتمد على زيادة حساسية كريات الدم الحمراء المعيبة للتكملة في ظل الظروف الأكثر ملاءمة من حيث تكوين الوسط هي اختبارات هيم (الحمضية) وهارتمان (مع السكروز).
يختبر كلا الاختبارين "بقاء" خلايا الدم الحمراء في عينة دم موضوعة في محلول ضعيف. يكون اختبار هيم إيجابيًا عندما يكون التدمير 5% أو أكثر، واختبار هارتمان 4% أو أكثر.
يتم إجراء اختبار كومبس لاستبعاد وجود اتصال مع آلية المناعة الذاتية لتدمير الخلايا، وهو سلبي بالنسبة للبيلة الهيموجلوبينية الليلية.

يشير تلوين البول إلى وجود نسبة كبيرة من الأوكسيهيموجلوبين فيه.
أظهر اختبار البول أن إحدى العلامات الأولية لمرض بيلة الهيموجلوبين الليلي هي بول الصباح والليل، ذو اللون الأحمر الداكن. مع مرور الوقت، ينفصل البول المتجمع إلى طبقات:
- السائل الموجود في الأعلى شفاف ولكنه يحتفظ باللون.
- يتم تحديد جزيئات الخلايا الميتة ذات الأصل العضوي من الأسفل.
ما هي الأمراض التي يجب التمييز بينها وبين بيلة الهيموجلوبين الليلية؟
يتم إجراء التشخيص التفريقي لبيلة الهيموغلوبين الليلي الانتيابي مع فقر الدم الآخر المماثل في المسار السريري، في المقام الأول مع فقر الدم الانحلالي من نوع المناعة الذاتية واللاتنسجي.
الميزات المشتركة هي:
- انخفاض حاد في عدد خلايا الدم الحمراء.
- كثرة الشبكيات.
- وجود اليرقان.
- حمى؛
- زيادة تركيز البيليروبين الحر.
- الميل إلى تجلط الدم.
- تضخم معتدل في الكبد والطحال.
في فقر الدم، لا توجد مستويات عالية من الهيموجلوبين في بلازما الدم واليوروبيلين في البول. تكون اختبارات هيم وهارتمان سلبية، لكن اختبار كومبس إيجابي.
يكون التشخيص صعبًا إلى حد كبير إذا حدث المرض في شكل أزمات مؤقتة على خلفية شكل حاد من سرطان الدم النقوي، وداء الكريات الحمر، وتصلب العظم والنقي، وآفات نخاع العظم النقيلي في الأورام الخبيثة.

يتم تخزين كتلة خلايا الدم الحمراء باردة في عبوات خاصة
علاج
حتى الآن، لا توجد وسيلة فعالة لوقف انهيار خلايا الدم الحمراء. كل ما تبقى هو استخدام خيار الاستبدال ونقل المريض بخلايا الدم الحمراء المغسولة من الجهات المانحة.
الميزة المهمة هي "الموقف" الجيد لجسم المريض تجاه الخلايا الأجنبية المحقونة، ولا يوجد عملياً أي رد فعل رفض. وبالنظر إلى وجود خلايا GPI-AP صحية في الأغشية وغياب الطفرات الجينية فيها، فمن الممكن دعم عملية تكون الدم لدى المريض.
يجب أن يبقى الدم المستخدم في نقل الدم مجمداً لمدة أسبوع على الأقل حتى يتم تدمير الكريات البيض الموجودة فيه بشكل كامل. بمجرد وصولها إلى المريض، يمكن أن تسبب تفاقم انحلال الدم بسبب زيادة الحساسية وتفعيل المكمل.
ومع عمليات نقل الدم المتكررة، لا يزال من الممكن تكوين الأجسام المضادة لكرات الدم الحمراء. في مثل هؤلاء المرضى، يتم إجراء عملية نقل الدم اللاحقة بعد عدة إجراءات لغسل خلايا الدم الحمراء بمحلول ملحي وفحص دم المتبرع باستخدام اختبار كومبس.
عادة ما يوصف عدد عمليات نقل الدم على الأقل خمس مرات، ولكن يعتمد ذلك على شدة حالة المريض واستجابته للعلاج.
لتحفيز تكوين الدم المناسب، يتم استخدام نيروبول (دواء هرموني ابتنائي) في دورات تصل إلى ثلاثة أشهر. في هذه الحالة، من الممكن حدوث تغيير في الحالة الوظيفية للكبد.
لغرض العلاج والوقاية من تكوين الخثرة، يتم استخدام الهيبارين، يليه الانتقال إلى جرعات الصيانة من مضادات التخثر غير المباشرة.
للتعويض عن فقدان الحديد، توصف أقراص.
قد يكون مؤشرا لإزالة الطحال زيادة حادة في الحجم وعلامات نوبة قلبية. نادرا ما يتم إجراء استئصال الطحال.
لحماية الكبد، توصف الأدوية الكبدية. في بعض الأحيان يساعد العلاج بالستيرويد.

يتم إعطاء الدواء عن طريق الوريد فقط
وفي السنوات الأخيرة، ظهرت معلومات عن استخدام عقار إيكوليزوماب (سوليريس)، المصنوع من الأجسام المضادة وحيدة النسيلة. إذا حكمنا من خلال التقارير المتاحة، فإنه يمنع انحلال الدم وقادر على مقاومة مكملات الدم. ويعتبر الدواء أغلى دواء في العالم. لم تتم دراسة تأثيره وآثاره السلبية بشكل كافٍ.
بيلة الهيموجلوبين الليلية ليس لها علاج محدد بعد. وحتى مع وجود رعاية داعمة كافية، يعيش المرضى حوالي خمس سنوات بعد ظهور المرض. لا يوجد منع. يجب على الجميع الالتزام بالسلوك الصحيح عند العمل والاتصال القسري بالمركبات السامة.
بيلة الهيموجلوبين الليلية الانتيابية، والمعروفة أيضًا باسم مرض ستروبينج-مارشيافافا، مرض مارشيافافا-ميشيلي، هو مرض نادر، وهو مرض دم تقدمي يهدد حياة المريض. وهو أحد أنواع فقر الدم الانحلالي المكتسب الناجم عن اضطرابات في بنية أغشية كرات الدم الحمراء. تتعرض الخلايا المعيبة للتحلل المبكر (انحلال الدم) الذي يحدث داخل الأوعية الدموية. هذا المرض وراثي بطبيعته، لكنه لا يعتبر وراثيا.
معدل الإصابة هو 2 حالة لكل مليون شخص. معدل الإصابة هو 1.3 حالة لكل مليون شخص سنويًا. يتجلى في الغالب في الأشخاص الذين تتراوح أعمارهم بين 25-45 سنة، ولم يتم تحديد أي اعتماد على الإصابة بالجنس والعرق. هناك حالات معزولة من المرض لدى الأطفال والمراهقين.
هام: متوسط العمر الذي يتم تشخيص المرض فيه هو 35 عامًا.
أسباب المرض
أسباب وعوامل الخطر لتطور المرض غير معروفة. لقد ثبت أن سبب المرض هو طفرة في جين PIG-A، الموجود في الذراع القصير للكروموسوم X. ولم يتم بعد تحديد العامل المطفر. في 30٪ من حالات بيلة الهيموجلوبين الانتيابي الليلي، هناك علاقة بمرض دم آخر - فقر الدم اللاتنسجي.
يحدث تكوين وتطور ونضج خلايا الدم (تكون الدم) في نخاع العظم الأحمر. تتكون جميع خلايا الدم المتخصصة مما يسمى بالخلايا الجذعية، وهي خلايا غير متخصصة احتفظت بالقدرة على الانقسام. تتشكل خلايا الدم الناضجة نتيجة للانقسامات والتحولات المتعاقبة، وتدخل مجرى الدم.
تؤدي طفرة في جين PIG-A حتى في خلية واحدة إلى تطور PNH. يؤدي تلف الجين أيضًا إلى تغيير نشاط الخلايا في عمليات الحفاظ على حجم نخاع العظم؛ حيث تتكاثر الخلايا الطافرة بشكل أكثر نشاطًا من الخلايا الطبيعية. في الأنسجة المكونة للدم، تتشكل بسرعة مجموعة من الخلايا التي تنتج خلايا دم معيبة. وفي هذه الحالة، فإن المستنسخ المتحور ليس ورمًا خبيثًا ويمكن أن يختفي تلقائيًا. يحدث الاستبدال الأكثر نشاطًا لخلايا نخاع العظم الطبيعية بخلايا متحولة في عمليات ترميم أنسجة نخاع العظم بعد حدوث أضرار كبيرة ناجمة، على وجه الخصوص، عن فقر الدم اللاتنسجي.
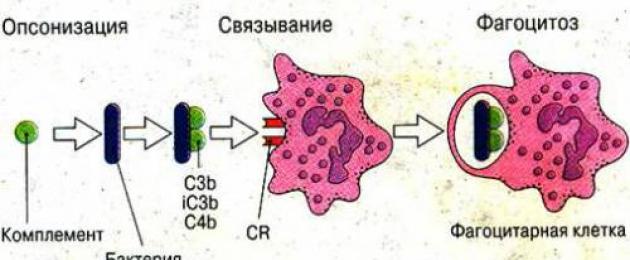
يؤدي تلف جين PIG-A إلى اضطرابات في تخليق بروتينات الإشارة التي تحمي خلايا الجسم من تأثيرات النظام المكمل. النظام المكمل عبارة عن بروتينات بلازما الدم المحددة التي توفر الحماية المناعية العامة. ترتبط هذه البروتينات بكريات الدم الحمراء التالفة وتذيبها، ويختلط الهيموجلوبين المتحرر مع بلازما الدم.
تصنيف
استنادا إلى البيانات المتاحة عن أسباب وخصائص التغيرات المرضية، يتم تمييز عدة أشكال من بيلة الهيموغلوبين الليلية الانتيابية:
- تحت الإكلينيكي.
- كلاسيكي.
- يرتبط باضطرابات تكون الدم.
غالبًا ما يسبق الشكل تحت الإكلينيكي للمرض فقر الدم اللاتنسجي. لا توجد مظاهر سريرية لعلم الأمراض، ولكن يتم اكتشاف وجود عدد صغير من خلايا الدم المعيبة فقط خلال الاختبارات المعملية.
في مذكرة. هناك رأي مفاده أن PNH هو مرض أكثر تعقيدا، والمرحلة الأولى منها هي فقر الدم اللاتنسجي.
يحدث الشكل الكلاسيكي مع أعراض نموذجية؛ حيث توجد مجموعات من خلايا الدم الحمراء والصفائح الدموية وبعض أنواع الكريات البيض المعيبة في دم المريض. تؤكد طرق البحث المختبرية تدمير الخلايا المتغيرة بشكل مرضي داخل الأوعية الدموية.
بعد المعاناة من الأمراض التي تؤدي إلى قصور تكوين الدم، يتطور شكل ثالث من الأمراض. تتطور الصورة السريرية الواضحة والتحلل داخل الأوعية لخلايا الدم الحمراء على خلفية آفات نخاع العظم.
هناك تصنيف بديل، وفقا لما يلي:
- في الواقع PNH، مجهول السبب.
- تتطور كمتلازمة مصاحبة لأمراض أخرى.
- تتطور نتيجة لنقص تنسج نخاع العظم.
لا ترتبط شدة المرض في الحالات المختلفة دائمًا بعدد خلايا الدم الحمراء المعيبة. تم وصف كل من الحالات تحت السريرية التي يصل فيها محتوى الخلايا المعدلة إلى 90%، والحالات الشديدة للغاية مع استبدال 10% من السكان الطبيعيين.
تطور المرض
من المعروف حاليًا أنه في دم المرضى الذين يعانون من بيلة الهيموجلوبين الليلية الانتيابية، يمكن أن توجد ثلاثة أنواع من كريات الدم الحمراء ذات حساسيات مختلفة للتدمير بواسطة النظام التكميلي. بالإضافة إلى الخلايا الطبيعية، تنتشر خلايا الدم الحمراء في مجرى الدم، وتكون حساسيتها أعلى بعدة مرات من المعدل الطبيعي. في دم المرضى الذين تم تشخيص إصابتهم بمرض مارشيافافا-ميشيلي، تم العثور على خلايا كانت حساسيتها للمكملات أعلى بـ 3-5 و15-25 مرة من المعدل الطبيعي.
تؤثر التغيرات المرضية أيضًا على خلايا الدم الأخرى، وهي الصفائح الدموية والخلايا المحببة. في ذروة المرض، يعاني المرضى من قلة الكريات الشاملة - وهو عدم كفاية عدد خلايا الدم من أنواع مختلفة.
تعتمد شدة المرض على النسبة بين مجموعات خلايا الدم السليمة والمعيبة. يتم تحقيق الحد الأقصى لمحتوى خلايا الدم الحمراء شديدة الحساسية لانحلال الدم المعتمد على المكملات خلال 2-3 سنوات من لحظة الطفرة. في هذا الوقت، تظهر الأعراض النموذجية الأولى للمرض.
يتطور المرض عادة بشكل تدريجي، ومن النادر ظهور الأزمة الحادة. تحدث التفاقم على خلفية الدورة الشهرية والإجهاد الشديد والأمراض الفيروسية الحادة والجراحة والعلاج بأدوية معينة (على وجه الخصوص، تلك التي تحتوي على الحديد). وفي بعض الأحيان يتفاقم المرض عند تناول أطعمة معينة أو بدون سبب واضح.
هناك أدلة على ظهور مظاهر مرض مارشيافافا-ميشيلي بسبب التشعيع.
يحدث بشكل مستمر انحلال خلايا الدم بدرجات متفاوتة في المرضى الذين يعانون من بيلة الهيموجلوبين الليلية الانتيابية المستمرة. تتخلل فترات التقدم المعتدل أزمات انحلالية، وتدمير هائل لخلايا الدم الحمراء، مما يؤدي إلى تدهور حاد في حالة المريض.
خارج الأزمة، يشعر المرضى بالقلق إزاء مظاهر نقص الأكسجة العام المعتدل، مثل ضيق التنفس، ونوبات عدم انتظام ضربات القلب، والضعف العام، وتفاقم القدرة على تحمل التمارين الرياضية. خلال الأزمة، تظهر آلام في البطن، موضعية بشكل رئيسي في منطقة السرة وفي أسفل الظهر. يتحول لون البول إلى اللون الأسود، ويكون الجزء الأكثر قتامة في الصباح. ولم يتم بعد تحديد أسباب هذه الظاهرة بشكل نهائي. مع PNH، يتطور عجين طفيف في الوجه، ويلاحظ اصفرار الجلد والصلبة.
مرض مارشيافافا-ميشيل، بيلة الهيموغلوبين الليلية الانتيابية مع بيلة هيموسيديرينية ثابتة، مرض ستروبينج-مارشيافافا هو نوع من فقر الدم الانحلالي المكتسب الذي يحدث مع انحلال الدم المستمر داخل الأوعية، بيلة هيموسيديرينية، تثبيط تكون الحبيبات والصفيحات.في ملاحظة! من الأعراض النموذجية للمرض هو البول الملون. في حوالي نصف الحالات المعروفة، لا يظهر المرض نفسه.
في الفترات الفاصلة بين الأزمات، قد يعاني المرضى من:
- فقر دم؛
- الميل إلى تجلط الدم.
- تضخم الكبد.
- مظاهر ضمور عضلة القلب.
- الميل إلى التهاب من أصل معدي.
عندما يتم تدمير خلايا الدم، يتم إطلاق مواد تزيد من تخثر الدم، مما يسبب تجلط الدم. قد تتشكل جلطات دموية في أوعية الكبد والكلى، كما تكون الأوعية التاجية والدماغية عرضة للتلف، مما قد يؤدي إلى الوفاة. يؤدي تجلط الدم الموضعي في أوعية الكبد إلى زيادة حجم العضو. تؤدي الاضطرابات في تدفق الدم داخل الكبد إلى تغيرات تنكسية في الأنسجة. عند انسداد نظام الوريد البابي أو الأوردة الطحالية، يتطور تضخم الطحال. تترافق اضطرابات استقلاب النيتروجين مع خلل في العضلات الملساء، ويشكو بعض المرضى من صعوبة في البلع، وتشنجات في المريء، ومن الممكن حدوث ضعف الانتصاب عند الرجال.
مهم! تؤثر مضاعفات التخثر في PNH في الغالب على الأوردة، ونادرا ما يتطور تجلط الدم.
فيديو - بيلة الهيموجلوبين الليلية الانتيابية
آليات تطور مضاعفات PNH
تتجلى الأزمة الانحلالية في الأعراض التالية:
- آلام البطن الحادة الناجمة عن تجلط الدم المتعدد في الأوردة المساريقية الصغيرة.
- زيادة اليرقان.
- ألم في منطقة أسفل الظهر.
- انخفاض ضغط الدم.
- زيادة درجة حرارة الجسم.
- تلطيخ البول باللون الأسود أو البني الداكن.
في حالات نادرة، تتطور "الكلية الانحلالية"، وهي شكل عابر محدد من الفشل الكلوي المصحوب بانقطاع البول الحاد. بسبب ضعف وظيفة الإخراج، تتراكم المركبات العضوية المحتوية على النيتروجين في الدم، وهي المنتجات النهائية لتحلل البروتين، ويتطور آزوتيميا. بعد تعافي المريض من الأزمة، يتم استعادة محتوى العناصر المشكلة في الدم تدريجيا، ويتلاشى اليرقان ومظاهر فقر الدم جزئيا.
المسار الأكثر شيوعا للمرض هو الأزمة، التي تتخللها فترات من الحالة المستقرة والمرضية. في بعض المرضى، تكون الفترات الفاصلة بين الأزمات قصيرة جدًا، وغير كافية لاستعادة تكوين الدم. يصاب هؤلاء المرضى بفقر الدم المستمر. هناك أيضًا نسخة مختلفة من الدورة مع بداية حادة وأزمات متكررة. وبمرور الوقت، تصبح الأزمات أقل تواترا. في الحالات الشديدة بشكل خاص، يكون الموت ممكنا، والذي يحدث بسبب الفشل الكلوي الحاد أو تجلط الأوعية الدموية التي تغذي القلب أو الدماغ.
مهم! لم يتم تحديد أي أنماط يومية في تطور الأزمات الانحلالية.
في حالات نادرة، يمكن أن يكون للمرض مسار هادئ طويل الأمد، وقد تم وصف حالات شفاء معزولة.
التشخيص
في المراحل المبكرة من المرض، يكون التشخيص صعبا بسبب ظهور أعراض غير محددة متفرقة. يتطلب التشخيص أحيانًا عدة أشهر من المراقبة. تظهر الأعراض الكلاسيكية - تلطيخ البول بشكل خاص - أثناء الأزمات وليس لدى جميع المرضى. أسباب الشك في مرض مارشيافافا-ميسيلي هي:
- نقص الحديد من مسببات غير معروفة.
- تجلط الدم والصداع ونوبات الألم في أسفل الظهر والبطن دون سبب واضح.
- فقر الدم الانحلالي من أصل غير معروف.
- ذوبان خلايا الدم، يرافقه قلة الكريات الشاملة.
- المضاعفات الانحلالية المرتبطة بنقل الدم الطازج من المتبرع.
في عملية التشخيص، من المهم إثبات حقيقة الانهيار المزمن لخلايا الدم الحمراء داخل الأوعية الدموية وتحديد العلامات المصلية المحددة للـ PNH.
في مجموعة من الدراسات، في حالة الاشتباه في بيلة الهيموجلوبين الانتيابية الليلية، بالإضافة إلى اختبارات البول والدم العامة، يتم إجراء ما يلي:
- تحديد محتوى الهيموجلوبين والهابتوغلوبين في الدم.
- النمط المناعي عن طريق قياس التدفق الخلوي لتحديد مجموعات الخلايا المعيبة؛
- الاختبارات المصلية، وخاصة اختبار كومبس.
التشخيص التفريقي مع بيلة الهيموجلوبين وفقر الدم لأسباب أخرى ضروري، على وجه الخصوص، ينبغي استبعاد فقر الدم الانحلالي المناعي الذاتي. الأعراض الشائعة هي فقر الدم واليرقان وزيادة البيليروبين في الدم. لا يلاحظ تضخم الكبد و/أو الطحال في جميع المرضى
علامات الانحلالي المناعي الذاتي
فقر دمبي إن جي اختبار كومبس + - زيادة المحتوى المجاني
الهيموجلوبين في بلازما الدم- + اختبار هارتمان (السكروز) - + اختبار هيم (الحمضية) - + الهيموسيدرين في البول - + تجلط الدم ± + تضخم الكبد ± ± تضخم الطحال ± ± نتائج اختبار هارتمان وهيم خاصة بالـ PNH وهي أهم العلامات التشخيصية.
علاج
يتم تخفيف الأزمة الانحلالية عن طريق عمليات نقل متكررة لخلايا الدم الحمراء أو إذابتها أو غسلها عدة مرات مسبقًا. ويعتقد أن هناك حاجة إلى 5 عمليات نقل دم على الأقل لتحقيق نتيجة دائمة، ومع ذلك، قد يختلف عدد عمليات نقل الدم عن المتوسط ويتم تحديده حسب شدة حالة المريض.
انتباه! ولا يمكن نقل الدم إلى هؤلاء المرضى دون تحضير مسبق. ونقل الدم المتبرع به يزيد من تفاقم الأزمة.
للتخلص من أعراض انحلال الدم، يمكن وصف نيروبول للمرضى، ولكن الانتكاسات ممكنة بعد التوقف عن تناول الدواء.
بالإضافة إلى ذلك، يتم وصف حمض الفوليك والحديد وأدوية حماية الكبد. عندما يتطور تجلط الدم، يتم استخدام مضادات التخثر ذات المفعول المباشر والهيبارين.
في حالات نادرة للغاية، تتم الإشارة إلى المريض لاستئصال الطحال - إزالة الطحال.
كل هذه التدابير داعمة، فهي تخفف من حالة المريض، ولكنها لا تقضي على عدد الخلايا الطافرة.
0

الأسباب:
ترتبط أسباب المرض بتدمير خلايا الدم الحمراء داخل الأوعية الدموية، والتي تكون معيبة إلى حد كبير. جنبا إلى جنب مع السكان المرضية لخلايا الدم الحمراء، يتم الحفاظ على بعض الخلايا الطبيعية التي لها عمر طبيعي. تم الكشف عن اضطرابات في بنية الخلايا المحببة والصفائح الدموية. المرض ليس وراثيا، ولكن أي عوامل خارجية تثير تكوين مجموعة من الخلايا المعيبة، وهي استنساخ، أي. لا يُعرف نسل خلية واحدة معدلة في البداية.ترتبط المضاعفات التخثرية في PNH بانحلال الدم داخل الأوعية، مما يؤدي إلى تكوين الخثرة. لا يزال أصل علامة مهمة ولكنها ليست إلزامية للمرض - نوبات بيلة الهيموجلوبين في الليل أو في الصباح - غير واضح.
لا ترتبط النوبة بالوقت من اليوم، بل بالنوم، والذي يمكن أن يسبب أيضًا أزمة أثناء النهار. هناك حساسية مكملة متزايدة لكريات الدم الحمراء المرضية في PNH. ولعل هذا هو الأساس لإثارة أزمة انحلال الدم عن طريق نقل الدم الطازج الذي يحتوي على عوامل تنشيط المتممة. نقل الدم المخزن لأكثر من أسبوع لا يسبب انحلال الدم.
أعراض بيلة الهيموجلوبين الليلية الانتيابية:
يتطور المرض ببطء: تظهر علامات فقر الدم المعتدل، والضعف، والتعب، والخفقان أثناء ممارسة الرياضة، وآلام في البطن، وغالبًا ما ترتبط بتجلط الأوعية المساريقية. يكون الجلد والأغشية المخاطية شاحبًا ويرقانيًا ورماديًا بسبب فقر الدم وترسب الهيموسيديرين. العلامات المميزة لانحلال الدم داخل الأوعية الدموية.ظهور البول الأسود ليس علامة دائمة. نظرًا لأن PNH غالبًا ما يكون مصحوبًا بنقص الكريات البيض (بسبب قلة المحببات بشكل رئيسي) ، فمن الممكن حدوث مضاعفات معدية مزمنة. قد يكون نقص الصفيحات معقدًا بسبب المتلازمة النزفية. يؤدي إفراز الهيموجلوبين والهيموسيديرين على المدى الطويل في البول تدريجيًا إلى تطور حالة نقص الحديد - تحدث متلازمة الوهن وجفاف الجلد وتظهر الأظافر الهشة.
تتميز صورة الدم في البداية بفقر الدم الطبيعي ثم فقر الدم الناقص الصباغ وكثرة الخلايا الشبكية الطفيفة (2-4٪ أو أكثر) ونقص الكريات البيض ونقص الصفيحات. مورفولوجية كريات الدم الحمراء ليس لها سمات مميزة. في النخاع العظمي يلاحظ تضخم الجرثومة الحمراء، أما في التريفين فيلاحظ زيادة طفيفة في خلوية النخاع العظمي، والتي قد تصبح ناقصة التنسج مع تقدم المرض.
بسبب انحلال الدم المستمر داخل الأوعية الدموية، يزداد محتوى الهيموجلوبين الحر في البلازما (عادة أقل من 0.05 جم / لتر). تكون مستويات الحديد في الدم طبيعية في البداية ولكنها قد تنخفض بشكل ملحوظ بعد ذلك. جنبا إلى جنب مع البداية النموذجية للمرض، عندما تسود متلازمة الانحلالي، قد تتطور صورة لمتلازمة اللاتنسجي، والتي يمكن أن تكون معقدة بعد بضع سنوات بسبب أزمة انحلالية مع بيلة هيموغلوبينية ليلية نموذجية. في كثير من الأحيان، تثير الأزمة الانحلالية نقل الدم.
تشخبص:
يتم التشخيص بناءً على علامات انحلال الدم داخل الأوعية الدموية (فقر الدم، كثرة الخلايا الشبكية الطفيفة، الهيموسيدرين في البول). يتم توضيح التشخيص من خلال دراسات خاصة (اختبار السكروز الإيجابي، اختبار هيم، اختبار كومبس السلبي).يحدث شكل الهيموليزين من فقر الدم الانحلالي المناعي الذاتي، المشابه في المظاهر الخارجية لـ PNH، مع انحلال الدم داخل الأوعية الدموية ويتميز بوجود الهيموليزينات في مصل الدم واختبار كومبس الإيجابي. على عكس PNH، لا يوجد نقص الكريات البيض أو نقص الصفيحات؛ عادة ما يكون للبريدنيزولون تأثير جيد. يمكن تمييز PNH عن فقر الدم اللاتنسجي من خلال صورة نخاع العظم: مع عدم تنسج تريبانات يتميز بغلبة الدهون، مع انحلال الدم - تضخم الخلايا، ومع ذلك، في حالات نادرة من PNH، يمكن أن تتطور صورة نقص تنسج نخاع العظم، على الرغم من اكتشاف الهيموسيديرين باستمرار في البول وكثرة الخلايا الشبكية في الدم.
علاج بيلة الهيموجلوبين الليلية الانتيابية:
لا يتم العلاج في حالة عدم وجود فقر الدم الشديد. تتطلب متلازمة فقر الدم الشديد نقل خلايا الدم الحمراء؛ يتم الحصول على أفضل النتائج عن طريق نقل كريات الدم الحمراء المغسولة أو القديمة لمدة 7-10 أيام. لنقص تنسج الدم، يشار إلى المنشطات: نيروبول - 10-20 ملغ يوميا أو ريتابوليل - 50 ملغ في العضل لمدة 2-3 أسابيع.يتم استخدام مكملات الحديد، لكنها قد تؤدي في بعض الأحيان إلى حدوث أزمة انحلالية. لمنع حدوث أزمة، يوصف الحديد بجرعات صغيرة أثناء العلاج بالستيرويدات الابتنائية. في حالة تجلط الدم، يشار إلى الهيبارين: في الحقنة الأولى، يتم إعطاء 10000 وحدة عن طريق الوريد، ثم 5-10 آلاف وحدة 2-3 مرات يوميًا تحت جلد البطن (بإبرة رفيعة على عمق 2 سم في المنطقة الدهنية). الأنسجة) تحت سيطرة تخثر الدم. موانع العلاج بالهيبارين هي التفاقم الأخير لقرحة المعدة أو الاثني عشر، وكذلك وجود مصادر النزيف.
بيلة الهيموجلوبين الليلية الانتيابية (PNH) هي مرض مكتسب يتجلى في فقر الدم الانحلالي المستمر، وبيلة الهيموجلوبين الانتيابية أو المستمرة، وانحلال الدم داخل الأوعية. تتميز ندرة هذا النوع من فقر الدم الانحلالي بحقيقة أن PNH يصيب شخصًا واحدًا من بين كل نصف مليون شخص، معظمهم من الشباب.
أسباب المرض غير معروفة حاليا. من المفترض أن يحدث ذلك بسبب حدوث استنساخ غير طبيعي لخلايا الدم الحمراء المعرضة لانحلال الدم داخل الأوعية الدموية. وفي المقابل، فإن نقص خلايا الدم الحمراء هو نتيجة للعيوب الهيكلية والكيميائية الحيوية في أغشيةها. ومن المعروف أن بيروكسيد الدهون يتم تنشيطه في الغشاء المعيب، مما يعزز التحلل السريع لخلايا الدم الحمراء، بالإضافة إلى ذلك، تشارك الحيوانات المستنسخة غير الطبيعية للخلايا المحببة والصفائح الدموية في العملية المرضية. الدور الرئيسي في حدوث مضاعفات التخثر من PNH ينتمي إلى تدمير خلايا الدم الحمراء داخل الأوعية الدموية وبدء تخثر الدم عن طريق العوامل التي تم إطلاقها خلال هذه العملية. PNH، كقاعدة عامة، يبدأ تدريجيا ويستمر بشكل مزمن مع الأزمات الدورية. تنجم الأزمات عن العدوى الفيروسية والتدخلات الجراحية والضغط النفسي والعاطفي والحيض واستخدام عدد من الأدوية والأطعمة.
أعراض بيلة الهيموجلوبين الليلية الانتيابية
أعراض PNH أثناء الأزمات:
- ألم الانتيابي في تجويف البطن.
- ألم في منطقة أسفل الظهر.
- يرقان الجلد والصلبة. ارتفاع الحرارة؛ عجين الوجه
- لون البول أسود، وخاصة في الليل.
- انخفاض حاد في ضغط الدم.
- تضخم عابر للطحال.
- توقف إخراج البول.
وفي بعض الحالات، تنتهي الأزمة الانحلالية بالوفاة.
أعراض PNH خارج الأزمة:
- ضعف عام؛
- لون البشرة شاحب مع لون اليرقان.
- فقر دم؛
- الميل إلى تجلط الدم. بول دموي؛ ضغط دم مرتفع؛ تضخم الكبد. ضيق التنفس؛ نبض القلب؛ الأمراض المعدية المتكررة.
التشخيص
- اختبار الدم: فقر الدم (طبيعي، ناقص الصباغ لاحقًا)، قلة الكريات البيض المعتدلة ونقص الصفيحات، ينخفض مستوى الحديد في الدم بشكل ملحوظ.
- فحص البول: تلطيخ أسود، بيلة هيموغلوبينية، بيلة هيموسيديرينية، بيلة بروتينية. اختبار البنزيدين في البول جريجيرسن إيجابي.
- اختبار هام المحدد إيجابي.
- اختبار هارتمان المحدد إيجابي.
- ثقب نخاع العظم: تضخم في السلالة الحمراء المكونة للدم، ولكن في الحالات الشديدة يمكن ملاحظة نقص تنسج نخاع العظم وزيادة في كمية الأنسجة الدهنية في نخاع العظم.
علاج بيلة الهيموجلوبين الليلية الانتيابية
علاج PNH هو عرضي ويتكون بشكل رئيسي من عمليات نقل الدم البديلة، والتي يعتمد حجمها وتكرارها على "الاستجابة" لهذه التدابير. في علاج PNH، يتم استخدام ميثاندروستينولون بجرعة 30-50 ملغ / يوم لمدة 2-3 أشهر على الأقل. تتم مكافحة نقص تنسج النخاع العظمي عن طريق الحقن الوريدي للجلوبيولين المناعي المضاد للخلايا الثيموسية بجرعة 150 ملغ / يوم لمدة 4 إلى 10 أيام. يوصى بتناول مكملات الحديد لكل نظام غذائي بجرعات صغيرة. في بعض الأحيان يكون للكورتيكوستيرويدات بجرعات عالية تأثير جيد. نقص تنسج النخاع العظمي مع تطور مضاعفات التخثر هي مؤشرات لزراعته. تم وصف حالات معزولة للشفاء من PNH، وفي بعض الحالات تكون مدة المسار الإيجابي للمرض عدة عقود.
الأدوية الأساسية
هناك موانع. مطلوب استشارة متخصصة.
فقر الدم اللاتنسجي هو مرض نادر في نظام الدم، يتميز بقلة الكريات الشاملة في الدم المحيطي ونخاع العظم تحت الخلايا (حتى عدم تنسج كامل) مع استبدال الأنسجة المكونة للدم النشطة بالأنسجة الدهنية. يعود الوصف الأول للمرض، الذي قدمه ب. إرليخ، إلى عام 1888.
يحدث المرض في معظم مناطق أوروبا وأمريكا بمعدل 2-3 حالات سنويًا لكل مليون نسمة. معدل الإصابة بفقر الدم اللاتنسجي أعلى بمقدار 2-3 مرات في شرق آسيا. هناك ذروتان لحدوث المرض: في سن 10 إلى 25 عامًا وفي الأشخاص الذين تزيد أعمارهم عن 60 عامًا، دون فروق ذات دلالة إحصائية حسب الجنس. الشكل النادر هو فقر الدم اللاتنسجي الخلقي - فقر الدم فانكوني، والذي يتجلى في معظم الحالات كمرض جسمي متنحي.
المسببات والتسبب في المرض
مسببات المرض في 70-80٪ من الحالات غير معروفة (أشكال مجهولة السبب)، وفي حالات أخرى يرتبط حدوث فقر الدم اللاتنسجي بعوامل كيميائية وفيزيائية مختلفة والتهابات (فقر الدم اللاتنسجي التالي لالتهاب الكبد، والأشكال المرتبطة بالفيروس المضخم للخلايا، عدوى فيروس البارفو، وما إلى ذلك).
الأكثر شيوعا هي الأشكال المكتسبة من فقر الدم اللاتنسجي، ولكن ما يصل إلى 15-20٪ من حالات المرض يمكن أن تكون متغيرات بنيوية / خلقية (فقر الدم فانكوني، فقر الدم المرتبط بخلل التقرن)، مصحوبة بالعديد من التشوهات الوراثية الخلوية. هناك أيضًا نوع مختلف من فقر الدم اللاتنسجي المرتبط ببيلة الهيموجلوبين الليلية الانتيابية.
الآلية المرضية الرئيسية لتطور عدم تنسج الدم في فقر الدم اللاتنسجي هي الضرر المناعي للخلايا الجذعية المكونة للدم. وفي الوقت نفسه، لا يمكن استبعاد وجود خلل وظيفي في الخلايا الجذعية المكونة للدم وأمراض البيئة الدقيقة المكونة للدم.
دليل على العمليات المناعية النشطة في نخاع العظام للمرضى الذين يعانون من فقر الدم اللاتنسجي هو زيادة في محتوى الخلايا اللمفاوية التائية الناضجة والمنشطة، والخلايا ذات النمط الظاهري الكابت القاتل، وانعكاس نسبة الكابت المساعد، والتي يتم اكتشافها بشكل طبيعي في هذه المجموعة من المرضى.
تتميز بزيادة في مستوى السيتوكينات التي تؤثر سلبا على عمليات المكونة للدم، مثل الإنترفيرون، IL-2، عامل نخر الورم (TNFα). في الوقت نفسه، يبدو أيضًا أن آلية الزناد المعززة غير المنضبطة لموت الخلايا المبرمج للخلايا المكونة للدم التي تعتمد على فاس تلعب دورًا مهمًا في تطور المرض. المرضى الذين يعانون من فقر الدم اللاتنسجي عادة لا يتميزون بنقص العوامل التي تنظم تكون الدم. هناك بعض الارتباطات المرضية بين فقر الدم اللاتنسجي، وبيلة الهيموجلوبين الليلية الانتيابية ومتلازمة خلل التنسج النقوي، والتي لا تزال طبيعتها غير واضحة تمامًا. يمكن أن يتحول فقر الدم اللاتنسجي في النهاية إلى بيلة هيموغلوبين ليلية انتيابية ومتلازمة خلل التنسج النقوي. تم اكتشاف استنساخ PNH صغير بدون علامات انحلال الدم، وفقًا للدراسات الحديثة، في 50-70٪ من المرضى الذين يعانون من فقر الدم اللاتنسجي. يمكن اكتشاف الحيوانات المستنسخة ذات التشوهات الوراثية الخلوية، في غياب الأدلة لصالح متلازمة خلل التنسج النقوي، لدى بعض المرضى الذين يعانون من فقر الدم اللاتنسجي.
الصورة السريرية
من حيث شدة الدورة، وارتفاع معدل الوفيات المبكرة بين المرضى الذين يعانون من فقر الدم اللاتنسجي الوخيم وتعقيد العلاج، فإن هذه الفئة قابلة للمقارنة مع مجموعة المرضى الذين يعانون من سرطان الدم الحاد. تصل نسبة الوفيات دون علاج في الأشهر الستة الأولى في الأشكال الشديدة من فقر الدم اللاتنسجي إلى 50٪ أو أكثر. أسباب وفاة المرضى هي تطور المرض وتطور المضاعفات المعدية النزفية والشديدة.
تنجم المظاهر السريرية للمرض بشكل رئيسي عن وجود متلازمة فقر الدم والنزف. يتميز المرضى الذين يعانون من فقر الدم اللاتنسجي بشحوب الجلد والأغشية المخاطية المرئية بدرجات متفاوتة من الشدة. كقاعدة عامة، هناك نزيف بأحجام مختلفة على الجلد والأغشية المخاطية - من الدقيق إلى المتموج. في كثير من الأحيان يكون هناك نزيف في قاع العين وشبكية العين، والذي يصاحبه انخفاض في حدة البصر. يمكن أن يكون النزيف على الأغشية المخاطية للتجويف الفموي مصحوبًا بأعراض التهاب الفم ونخر الأنسجة الرخوة. في الأشكال الشديدة من المرض مع المظاهر النزفية الشديدة، من الممكن حدوث نزيف في جدار الأمعاء. في الحالة الأخيرة، ستحدث الصورة السريرية المقابلة: الألم والانتفاخ والألم عند الجس، واضطرابات التمعج. في الوقت نفسه، في بعض المرضى (في المتوسط ما يصل إلى 20٪)، لم يتم ملاحظة أي مظاهر نزفية مرئية أثناء الفحص الأولي. تتجلى التغييرات في نظام القلب والأوعية الدموية من خلال عدم انتظام دقات القلب، وتوسيع حدود القلب، وأصوات القلب المكتومة،. والنفخة الانقباضية فوق سطح القلب.
اعتلال العقد اللمفية وتضخم الكبد والطحال ليست نموذجية لفقر الدم اللاتنسجي. مع قلة المحببات العميقة، هناك ميل متزايد لتطوير مضاعفات معدية والتهابية نخرية.
لوحظ ظهور حاد لفقر الدم اللاتنسجي في 12-15٪ من المرضى ويصاحبه حمى والتهاب الحلق الناخر ونزيف حاد في الأنف واللثة والرحم وظهور نزيف متعدد على الجلد والأغشية المخاطية. في أكثر من 80٪ من المرضى، يتطور المرض تدريجيا مع زيادة مظاهر فقر الدم والمتلازمة النزفية.
مع فقر الدم فانكوني، والذي يتم اكتشافه عادة في سن مبكرة، يمكن اكتشاف تشوهات الهيكل العظمي وتصبغ الجلد - بقع القهوة بالحليب.
البحوث المختبرية
يُظهر تعداد الدم الكامل عادةً قلة الكريات الشاملة مع الحفاظ النسبي على الخلايا الليمفاوية. فقر الدم عادة ما يكون طبيعي اللون ويتميز بقلة الخلايا الشبكية. يمكن ملاحظة كثرة الكريات الكبيرة. يقل عدد الصفائح الدموية بشكل كبير وعادةً ما يكون حجمها صغيرًا.
تتميز صورة النخاع العظمي للمرضى الذين يعانون من فقر الدم اللاتنسجي بانخفاض عدد الخلايا المكونة للدم وتضخم المساحات الدهنية. غالبًا ما يتم ملاحظة خلل تكون الكريات الحمر أو غيابه ؛ وهو ما لا يصاحبه تغيرات خلل التنسج في سلسلة أخرى من تكون الدم ، كما هو الحال في متلازمة خلل التنسج النقوي. يتم تقليل عدد الخلايا المكروية والخلايا المحببة بشكل كبير. نظرًا لأن تلف نخاع العظم غير متساوٍ، فقد يتم ملاحظة تضخم بؤري في سلالات الكريات الحمر والخلايا المحببة، وعند سحب "الجيب الساخن" مع تركيز تكون دموي سليم، قد تكون مؤشرات الميلوجرام، خاصة في المراحل المبكرة من المرض، قريبة إلى وضعها الطبيعي. لتقييم الخلوية الشاملة وتقييم مورفولوجية الخلايا المكونة للدم المتبقية، دراسة عينة خزعة نخاع العظم عالية الجودة من تريفين أمر بالغ الأهمية.
تشخيص متباين
يتم تشخيص فقر الدم اللاتنسجي بناءً على تحديد قلة الكريات الشاملة في الدم المحيطي وانخفاض خلوية نخاع العظم وفقًا لخزعة التريبانوبيا. السمة هي استبدال الأنسجة المكونة للدم النشطة بالأنسجة الدهنية، في غياب تسلل الخلايا غير النمطية وعلامات التليف. يسمح لنا الفحص الدقيق لمسحات الدم ومستحضرات نخاع العظم باستبعاد وجود العدلات خلل التنسج والصفائح الدموية غير الطبيعية والخلايا السرطانية.
عند تشخيص فقر الدم اللاتنسجي، توصي مجموعات البحث الدولية بمراعاة وجود اثنين على الأقل من مؤشرات الدم التالية مع التغيرات المميزة في صورة نخاع العظم: مستوى الهيموجلوبين
تشتمل خطة الفحص للمرضى الذين يشتبه في إصابتهم بفقر الدم اللاتنسجي على فحص دم سريري كامل مع تحديد عدد الصفائح الدموية والخلايا الشبكية، وتعداد الميلوجرام والفحص النسيجي لخزعة من تريفين نخاع العظم. من أجل تحديد المتغيرات المرضية المرتبطة بوجود استنساخ PNH، يُنصح جميع المرضى الذين يعانون من فقر الدم اللاتنسجي باختبار خلايا الدم بحثًا عن بيلة هوموجلوبينية ليلية انتيابية باستخدام قياس التدفق الخلوي عالي الحساسية. يخضع المتلقون المحتملون لنخاع العظم إلى كتابة HLA لخلايا الدم.
لتشخيص الأشكال الخلقية النادرة للمرض، من المهم الحصول على تاريخ شامل وفحص المريض. من أجل استبعاد فقر الدم فانكوني، يشار إلى تحليل الكروموسومات للخلايا الليمفاوية في الدم - اختبار لانهيار الكروموسومات المستحثة مع ديبوكسي بيوتان أو ميتوميسين.
عند إجراء التشخيص التفريقي، من الضروري استبعاد قلة الكريات من أصل ثانوي. هذا، بالإضافة إلى التاريخ التفصيلي والفحص، قد يتطلب إجراء اختبارات مثل تحديد مستوى فيتامين ب 12 وحمض الفوليك في الدم، والاختبارات الفيروسية، والتنميط المناعي لخلايا نخاع العظم، والموجات فوق الصوتية وتخطيط صدى القلب، واختبارات استبعاد أمراض الروماتويد وغيرها من الاختبارات مثل مبين.
يتم إجراء التشخيص التفريقي أيضًا مع عدم تنسج الخلايا الحمراء الجزئي المكتسب والشكل الخلقي - فقر الدم Diamond-Blackfan، حيث يتم اكتشاف عدم تنسج سلالة الكريات الحمر لنخاع العظم مع الحفاظ على الحبيبات والصفيحات.
تصنيف
لتحديد أساليب العلاج، من الضروري تحديد شدة فقر الدم اللاتنسجي. وفقا للتصنيف الدولي، من المعتاد التمييز بين الأشكال الشديدة وغير الشديدة من فقر الدم اللاتنسجي. كان الغرض الرئيسي من هذا التصنيف هو تحديد مجموعة من المرضى الذين هم في المقام الأول مرشحون لزراعة نخاع العظم بسبب خطر الوفاة المبكرة.
علاج
يجب أن تهدف استراتيجية علاج فقر الدم اللاتنسجي إلى استعادة النقص في الخلايا الجذعية المكونة للدم وقمع العمليات المناعية المدمرة.
لا يمكن تحقيق الاستعادة الكاملة لتكوين الدم في نخاع العظم لدى المرضى الذين يعانون من فقر الدم اللاتنسجي إلا من خلال زرع الخلايا الجذعية المكونة للدم، وهي الطريقة المفضلة لدى المرضى الشباب الذين يعانون من أشكال حادة وفائقة الخطورة من المرض. ومع ذلك، فإن الطريقة الرئيسية للعلاج بالنسبة لغالبية المرضى هي العلاج المثبط للمناعة باعتباره وسيلة أكثر سهولة، مع موانع أقل وقابلة للمقارنة في الفعالية مع زرع الخلايا الجذعية المكونة للدم.
تمت المحاولات الأولى لعلاج فقر الدم اللاتنسجي عن طريق زرع نخاع العظم في ثلاثينيات القرن العشرين، لكن التعقيد والعيوب في تكنولوجيا اختيار المتبرعين وتقنيات زرع الأعضاء في ذلك الوقت حدت من إمكانيات استخدام زرع الأعضاء. مع تحسن تقنيات وطرق اختيار المتبرعين، أصبح زرع نخاع العظم جزءًا من العلاج القياسي للمرضى الذين يعانون من فقر الدم اللاتنسجي الوخيم كطريقة اختيار للمرضى الذين تم تشخيصهم حديثًا والذين يعانون من فقر الدم اللاتنسجي الوخيم في وجود متبرع ذي صلة متطابق مع HLA وك طريقة لعلاج المرضى الذين يعانون من مرض شديد والذين لم يستجيبوا للعلاج بالجلوبيولين المناعي المضاد للخلايا الثيموسية والسيكلوسبورين. تم تحقيق زيادة كفاءة زراعة نخاع العظم الخيفي نتيجة لتقليل حدوث المضاعفات المعدية، وتحسين أنظمة التحضير قبل الزرع، وتقليل حدوث تفاعلات الرفض ومرض الكسب غير المشروع مقابل المضيف.
وفقًا لمجموعة العمل الأوروبية المعنية بزراعة نخاع العظم وفقر الدم اللاتنسجي، كان معدل البقاء على قيد الحياة للمرضى الذين يعانون من فقر الدم اللاتنسجي الوخيم بعد زرع الخلايا الجذعية المكونة للدم في 1970-1979. 43% في الفترة 1991-1996 ارتفعت إلى 69%، وبحلول الفترة 1997-2002. - ما يصل إلى 72٪. يمكن أن يصل معدل البقاء على قيد الحياة على المدى الطويل للمرضى الذين يعانون من فقر الدم اللاتنسجي بعد عملية الزرع إلى 80-96٪. المصدر المفضل للخلايا الجذعية المكونة للدم للمرضى الذين يعانون من فقر الدم اللاتنسجي هو نخاع العظام.
في المرضى الذين يعانون من فقر الدم اللاتنسجي الخفيف وفقر الدم اللاتنسجي الوخيم والذين تزيد أعمارهم عن 40 عامًا و/أو ليس لديهم متبرع شقيق متطابق مع HLA، يوصى بإجراء دورة من العلاج المثبط للمناعة. يعتمد استخدام العلاج المثبط للمناعة على مفهوم التسبب في فقر الدم اللاتنسجي كعملية مرضية ناجمة عن انتهاك التنظيم المناعي لتكوين الدم. النظام القياسي للعلاج المثبط للمناعة، والذي يعطي أفضل النتائج للمرضى الذين يعانون من فقر الدم اللاتنسجي الوخيم وفقر الدم اللاتنسجي غير الشديد، هو مزيج من الغلوبولين المناعي المضاد للخلايا التوتية والسيكلوسبورين أ. وقد تم تأكيد مزايا العلاج المشترك من قبل العديد من المجموعات البحثية. وهكذا، أظهرت نتائج العلاج المثبط للمناعة لمدة 11 عامًا، وفقًا لمجموعة من العلماء الألمان، زيادة في تكرار حالات التعافي عند إضافة الغلوبولين المناعي المضاد للخلايا الثيموسية والسيكلوسبورين إلى العلاج من 41 إلى 70٪ في المجموعة العامة من المرضى ومن 31 إلى 70٪ في المجموعة العامة من المرضى ومن 31 إلى 70٪ في المجموعة العامة من المرضى. 65% في حالات فقر الدم اللاتنسجي الشديد. وفي الوقت نفسه، انخفض متوسط الوقت اللازم لتحقيق التعافي من 82 إلى 60 يومًا، وزاد معدل الإصابة بالانتكاس بنسبة 18%.
الغلوبولين المناعي المضاد للخلايا الثيموسية هو دواء يتم الحصول عليه عن طريق تحصين الحيوانات بالخلايا الليمفاوية البشرية (الخلايا الثيموسية الجنينية). الأدوية من هذه السلسلة لها تأثير انتقائي سام للخلايا اللمفاوية ضد مثبطات T المنشطة، وتمنع إنتاج السيتوكينات القمعية بواسطة الخلايا التائية، وتعمل على موت الخلايا المبرمج عن طريق تقليل التعبير عن مستضد Fas على خلايا CD+ في نخاع العظم لدى المرضى.
السيكلوسبورين A هو مستقلب من فطر Tolipocladium inflatum، وهو عديد ببتيد حلقي يغير وظيفة الخلايا الليمفاوية بشكل انتقائي وعكسي، مما يمنع إنتاج وتثبيت الليمفوكينات على مستقبلات محددة؛ يثبط مرحلتي G0 وG1 من دورة الخلية للخلايا ذات الكفاءة المناعية، ويقلل من نشاط الجينات المسؤولة عن تخليق IL-2 وعدد من السيتوكينات الأخرى. تتمثل ميزة CsA في تأثيرها العكسي المحدد في حالة عدم وجود تأثير قمعي على تكون الدم، فضلاً عن الحفاظ النسبي على المناعة المضادة للعدوى.
يتم إجراء دورات العلاج بالجلوبيولين المناعي المضاد للخلايا الثيموسية، لمدة 4-5 أيام، في المستشفى. الجرعات الموصى بها من دواء الغلوبولين المناعي المضاد للخلايا الثيموسية للخيول هي 20-40 ملغم / كغم من وزن الجسم. لتحسين النتائج ومنع ردود الفعل التحسسية ومرض المصل، عادة ما يتم وصف الجلايكورتيكويدات في وقت واحد في شكل دورة قصيرة [ميثيل بريدنيزولون بجرعة 1-3 ملغم / كغم)]. بعد الانتهاء من إعطاء الغلوبولين المناعي المضاد للخلايا الثيموسية، توصف مستحضرات CsA عن طريق الفم لفترة طويلة (من 6 أشهر) بجرعات تتراوح بين 5-7 ملغم / كغم وأعلى في حالة عدم وجود سمية كبيرة. عند استخدام هذا النظام، يكون معدل الاستجابة 60-80%، مع معدل البقاء على قيد الحياة لمدة 5 سنوات للمرضى الذين يعانون من فقر الدم اللاتنسجي الوخيم 75-85%.
عادة ما يتم ملاحظة النتائج الإيجابية المستمرة الأولى أثناء العلاج المثبط للمناعة بعد 2-3 أشهر، ولذلك فمن المستحسن تحديد نتائج العلاج بعد 3-6 أشهر من بداية العلاج. معايير فعالية العلاج هي مغفرة كاملة وجزئية. مغفرة سريرية ودموية كاملة تعني عدم وجود أعراض سريرية للمرض، وتخفيف كامل لمتلازمة النزف، ومحتوى الهيموجلوبين أكثر من 110 جم / لتر؛ محتوى المحببات أكثر من 1.0x109 / لتر، محتوى الصفائح الدموية أكثر من 100x109 / لتر (في حالات أخرى - أكثر من 125-150x109 / لتر). تتميز المغفرة السريرية والدموية الجزئية بغياب الأعراض السريرية للمرض ومظاهر المتلازمة النزفية، ومحتوى الهيموجلوبين أكثر من 80 جم / لتر مع الاستقلال عن العلاج بمكونات الدم، ومحتوى المحببات أكثر من 0.5 × 109 / لتر، والصفائح الدموية. أكثر من 20.0x109/لتر.
قد تكون النتيجة الإيجابية أيضًا تحسنًا سريريًا ودمويًا، حيث لا توجد مظاهر نزفية واضحة، وتقل الحاجة إلى العلاج بمكونات الدم وهناك تحسن في مؤشرات الدم مع محتوى محبب يزيد عن 0.5x10 9/ لتر، والصفائح الدموية أكثر من 20.0x109/لتر.
لتقييم فعالية العلاج للمرضى الذين يعانون من فقر الدم اللاتنسجي اعتمادا على شدة المرض، يقترح فريق الخبراء الأوروبي المعايير التالية. وفقًا للتوصيات الحديثة، يجب أن يستمر CsA بعد الحصول على الحد الأقصى من الاستجابة الدموية [مغفرة جزئية مستدامة مع تحسن في جميع الخطوط المكونة للدم، مغفرة كاملة] لمدة 6 إلى 12 شهرًا، يليها الانسحاب التدريجي، مما يقلل من عدد الانتكاسات.
هناك تجربة إيجابية مع استخدام جرعات عالية من السيكلوفوسفاميد في الخط الأول من العلاج. أظهرت المنشورات الأولى التي يرجع تاريخها إلى عام 1996 تأثيرًا جيدًا للعلاج المثبط للمناعة بهذه الأدوية لدى المرضى الذين يعانون من فقر الدم اللاتنسجي، ولكن مع مضاعفات خطيرة أثناء العلاج، بما في ذلك الالتهابات المميتة. ومع ذلك، مع تحسن العلاج المصاحب، تظهر المنشورات الحديثة نتائج علاجية جيدة مع حالات هدأة أكثر اكتمالًا واستدامة في المرضى الذين يعانون من فقر الدم اللاتنسجي الوخيم، على الرغم من أن هذه النتائج لم يتم تأكيدها من خلال تجارب عشوائية محكومة.
إذا كانت الدورة الأولى من العلاج المركب مع الغلوبولين المناعي للخلايا الثيموسية / CsA غير فعالة للمرضى الذين يعانون من فقر الدم اللاتنسجي الوخيم، يتم النظر في إمكانية زرع نخاع العظم من متبرع متوافق غير ذي صلة. علاوة على ذلك، فإن احتمالية الحصول على نتائج إيجابية تكون أعلى عند إجراء عملية الزرع في وقت مبكر.
تشمل عيوب العلاج المثبط للمناعة كوسيلة لعلاج المرضى الذين يعانون من فقر الدم اللاتنسجي ما يلي:
الحفاظ على عيوب المكونة للدم المتبقية (في شكل الحفاظ على بؤر نقص تنسج نخاع العظم، والنقص الوظيفي للخلايا النقوية)؛
ارتفاع خطر الانتكاس (ما يصل إلى 20-30٪ من المرضى وما فوق)؛
المضاعفات النسيلية المتأخرة (تصل إلى 20-60٪ مع المراقبة على المدى الطويل)، بما في ذلك متلازمة خلل التنسج النقوي، وسرطان الدم الحاد، وبيلة الهيموجلوبين الليلية الانتيابية.
إن تواتر الانتكاسات لدى المرضى الذين يعانون من فقر الدم اللاتنسجي بعد الخط الأول من العلاج المثبط للمناعة مرتفع نسبيًا، ولكن في معظم الحالات يتم علاج هذه الانتكاسات بنجاح من خلال دورات متكررة من العلاج المثبط للمناعة ولا تؤدي إلى تفاقم التشخيص العام بشكل ملحوظ. وهكذا، أظهرت الدراسات التي أجريت في السنوات الأخيرة أنه في حالة الانتكاس بعد أول دورة علاجية ناجحة، والتي تشمل الغلوبولين المناعي المضاد للخلايا التوتية، فإن الدورات المتكررة تؤدي إلى هدأة في 11-65٪ من المرضى.
في الخط الثاني والخطوط اللاحقة من العلاج، من الممكن استخدام أدوية مثل alemtuzumab ومستحضرات حمض الميكوفينوليك لعدم تحمل CsA. هناك أدلة على تجربة إيجابية مع استخدام عقار داكليزوماب (الأجسام المضادة وحيدة النسيلة المؤتلفة ضد مستقبل IL-2) وعدد من الأدوية المثبطة للمناعة الأخرى، ولكن لا توجد حتى الآن بيانات مقنعة بما فيه الكفاية عن استخدامها في مجموعات كبيرة من المرضى الذين يعانون من عدم التنسج. فقر دم.
نادرًا ما يتم الآن استخدام استئصال الطحال، الذي كان يستخدم سابقًا في علاج المرضى الذين يعانون من فقر الدم اللاتنسجي، على الرغم من أن بعض المؤلفين يعتبرون استخدامه مبررًا في الخط الثاني والثالث من العلاج، خاصة في وجود مكون مناعة ذاتية.
لقد ثبت أن البدء المبكر في دورة العلاج والعلاج المصاحب المناسب لهما أهمية كبيرة في تحسين نتائج علاج فقر الدم اللاتنسجي. ويتضمن الأخير العلاج ببدائل الدم للحفاظ على مستوى خلايا الدم الحمراء والصفائح الدموية عند مستوى آمن.
مؤشرات لاستخدام تركيز الصفائح الدموية هي متلازمة النزفية مع مستويات الصفائح الدموية
في السنوات الأخيرة، جرت محاولات لاستخدام منبهات مستقبلات الثرومبوبويتين (الترومبوباج) للسيطرة على المتلازمة النزفية، وقد أظهرت هذه المحاولات نتائج جيدة. علاوة على ذلك، هناك بيانات تشير إلى قدرة منبهات مستقبلات الثرومبوبويتين على أن تؤدي ليس فقط إلى زيادة عدد الصفائح الدموية وتخفيف المظاهر النزفية، ولكن أيضًا إلى تحسين خطوط الخلايا الأخرى.
نظرًا لأن الاعتماد على نقل الدم غالبًا ما يؤدي إلى زيادة الحديد بعد نقل الدم لدى المرضى الذين يعانون من فقر الدم اللاتنسجي، فإن المرضى الذين يعانون من عمليات نقل خلايا الدم الحمراء المتكررة ومستويات الفيريتين في الدم أكبر من 1000 ملغم / لتر يتم وصفهم للعلاج بمخلب الحديد.
في حالة حدوث مضاعفات معدية لدى مرضى فقر الدم اللاتنسجي، يتم العلاج وفقًا للقواعد الشائعة للمرضى الذين يتلقون العلاج المثبط للمناعة، مع وصف الأدوية المضادة للبكتيريا واسعة الطيف والأدوية المضادة للفطريات كما هو محدد.
لا يعتبر معظم الباحثين استخدام المنشطات المكونة للدم - عامل تحفيز مستعمرة الخلايا المحببة والإريثروبويتين - في المرضى الذين يعانون من فقر الدم اللاتنسجي أمرًا مستحسنًا بسبب فعاليتها المنخفضة في هذه المجموعة من المرضى وزيادة خطر الإصابة بمضاعفات نسيلية. أظهرت البيانات المستقاة من الملاحظات طويلة المدى والتحليل التلوي، والتي تم تقديمها بانتظام في العقد الأول من القرن الحادي والعشرين في المؤتمرات العلمية ومؤتمرات أمراض الدم للجمعية الأمريكية لأمراض الدم، والجمعية الأوروبية لأمراض الدم، والمجموعة الأوروبية لزراعة نخاع العظام وغيرها، أن استخدام الإريثروبويتين ولا يؤثر G-CSF بشكل كبير على تقليل معدل الوفيات أو زيادة الاستجابات الكاملة والشاملة للعلاج. ومع ذلك، قد يوصى باستخدام دورات قصيرة من G-CSF للعدوى الجهازية الشديدة لدى المرضى الذين يعانون من قلة المحببات العميقة. يعتمد تشخيص المرض بشكل أساسي على شدة عدم تنسج الدم والبدء المبكر للعلاج النشط. بدون علاج، في أشكال حادة، يموت ما يصل إلى 50٪ من المرضى في الأشهر الأولى، ومع العلاج الحديث، البقاء على قيد الحياة على المدى الطويل هو 70-80٪.
أما بالنسبة للعلاج المثبط للمناعة، فإن نتائج العلاج تكون أفضل لدى المرضى الذين يعانون من استجابة مبكرة للخلايا المحببة والشبكية. تشير البيانات المتاحة أيضًا إلى استجابة أفضل للعلاج المثبط للمناعة لدى المرضى الذين يعانون من فقر الدم اللاتنسجي المرتبط بوجود استنساخ PNH. من بين العوامل التي تؤثر على تشخيص المرض فعالية العلاج المثبط للمناعة واحتمال التطور النسيلي، وقد تم لفت الانتباه مؤخرًا إلى تقصير طول التيلومير في خلايا الدم.






