Фенилкетонурия представляет собой достаточно тяжелое наследственное заболевание, основная тяжесть проявлений которого сосредоточена, прежде всего, на нервной системе. Фенилкетонурия, симптомы которой чаще всего встречаются среди девочек, возникает по причине нарушения обмена аминокислот, учитывая же поражение при этом ЦНС, ее проявления сводятся к нарушению умственного развития.
Общее описание
В классической своей форме, которая актуальна для большинства случаев, фенилкетонурия, которую также принято определять как фенилпировиноградную олигофрению, связана с резкостью снижения активности печеночного фермента. Им в частности является фениланин-4-гидроксилаз. Нормальное состояние организма предусматривает катализацию им превращения в тирозин фенилаланина. 1% случаев отмечается возникновением атипичной формы фенилкетонурии, которая возникает по причине мутаций в другого типа генах, которые ответственны за процесс кодирования ферментов. Наследование заболевания происходит в соответствии с аутосомно-рецессивной схемой.
Что касается процессов, происходящих при рассматриваемом заболевании, то они заключаются в следующем. Возникновение характерного метаболического блока провоцирует активацию побочных путей по обмену фенилаланина, что приводит к накоплению токсичных производных от его действия. К таким производным в частности относятся фенилмолочная и фенилпировиноградные кислоты, практически не образующиеся при нормальном состоянии организма. Они же влияют на ЦНС, провоцируя нарушения белкового обмена, обмена липопротеидов и обмена гликопротеидов. Одновременно с этим возникают расстройства в транспорте аминокислот, нарушения в обмене серотонина и катехоламинов, актуальность приобретают также и перинатальные факторы.
Помимо этого начинают также образовываться и в норме практически отсутствующие ортофенилацетат и фенилэтиламин. Их избыток провоцирует нарушения в метаболизме липидов, происходящем в головном мозге. Как предполагается, именно это становится причиной прогрессирующего состояния снижения у больных данным заболеванием интеллекта, что может достичь даже идиотии. В целом же окончательно механизм, в соответствии с действием которого происходит нарушение в развитии функций мозга в случае с актуальным для организма заболеванием фенилкетонурией, не ясен.
Фенилкетонурия: симптомы заболевания
Здесь, прежде всего, важно отметить, что первые недели жизни ребенка не позволяют внешне определить данного заболевания. Первые его признаки проявляются через два-шесть месяцев с момента рождения малыша. Он становится вялым, наблюдается отсутствие заинтересованности в отношении условий, его окружающих и мира в целом. Также ребенок становится беспокойным, нарушениям подвергается мышечный тонус. Появляется рвота, судороги, тяжелые кожные . Под экземами в частности подразумевается острая или хроническая форма воспалительного и незаразного заболевания, при котором образуется сыпь. Природа заболевания аллергическая, дополнительными проявлениями симптоматики выступает чувство жжения и выраженный зуд кожи. Актуальна и склонность к рецидивам, то есть к повторному возникновению симптоматики после относительного временного его затишья.
Шестой месяц позволяет определить отставание в развитии у ребенка. Одновременно с этим теряется способность к фокусированию взгляда на отдельных предметах, малыш перестает узнавать родителей. Отсутствует реакция на цветные/яркие игрушки. Важно оперативно приступить к лечению, в противном случае отсталость развития будет постепенно подвергаться лишь прогрессированию в актуальных для нее процессах.
Физическое развитие больных младенцев на физическом уровне отмечается меньшими нарушениями, нежели на психическом. В обхвате голова может быть несколько меньших размеров, чем это предусмотрено для показателей нормы. Зубки прорезываются позже, позже ребенок начинает сидеть и ходить. Принятие положения стоя у таких малышей сопряжено с широким расставлением для этого ног, а также со сгибанием их в коленях и в тазобедренных суставах, плечи и голова при этом опущены. Что касается особенностей ходьбы у больных детей, то она характеризуется покачиваниями, шажки небольшие. Сидят дети, поджав под себя ножки, что обуславливается значительным мышечным тонусом, который они испытывают.
Отличаются детки и характерной внешностью со светлыми волосами, кожа у них абсолютно белая, без пигментации, глаза светлые. Учитывая излишнюю белизну кожи, она нередко покрывается у детей сыпью, что объясняется ее особой чувствительностью в отношении воздействия ультрафиолетового излучения.
В числе основных проявлений, свойственных фенилкетонурии, можно также выделить характерный «мышиный» запах, в некоторых случаях возможны , которые, однако, с возрастом исчезают. Выраженными проявлениями выступают синюшность конечностей, дермографизм (изменение в окраске кожи местного типа, происходящее при механическом воздействии на нее), потливость. Чаще среди больных фенилкетонурией отмечается, помимо перечисленных симптомов, наличие , частых запоров, тремор (т.е. дрожание), потеря равновесия и расстройство в виде нарушения координации движений.
Диагностирование фенилкетонурии
Важным, как мы уже отметили, является раннее диагностирование заболевания, что позволит избежать его развития и привести к ряду необратимых и тяжелых последствий. По этой причине в родильных домах к 4-5 дням жизни (для новорожденных доношенных) берется для анализа кровь. У недоношенных детей на предмет фенилкетонурии (ФКУ) кровь берется на 7 день.
Процедура предусматривает взятие капиллярной крови по прошествии часа с момента кормления, ею в частности пропитывается специальный бланк. Концентрация, указывающая на отметку свыше 2,2% фенилаланина в крови малыша, требует направления его с родителями для осмотра в медико-генетический центр. Там же проводится дообследование и, собственно, уточнение диагноза.
Причины фенилкетонурии
Фенилкетонурия может быть спровоцирована следующими факторами:
- Близкородственные браки, при которых, помимо иных патологий, повышается вероятность рождения ребенка с этим заболеванием;
- Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам в области локализации 12 хромосомы.

Лечение фенилкетонурии
Единственный метод лечения заключается в своевременной организации диетотерапии, которая требуется с первых дней жизни. Заключается она в резком ограничении фенилаланина, который содержится в отдельных пищевых продуктах. Таким образом, исключаются все белковые продукты. Учитывая продолжительность и полное исключение фенилаланина из пищи, возможным становится расщепление собственных белков, что ведет к истощению организма больного. По этой причине потребность в получении белка возмещается за счет аминокислотных смесей и белковых гидролизатов.
Со сменой снижения концентрации в крови фенилаланина до нормальных показателей, в рацион постепенно добавляются продукты животного происхождения. В рацион добавляются различные фрукты и сезонные овощи, растительные и животные жиры, а также углеводы. Естественно, что контроль над содержанием фенилаланина следует продолжать вести. Организм должен быть снабжен данной аминокислотой в должном объеме для обычного развития и роста малыша, однако, за исключением возможности скапливания его в тканях.
Строжайшая диета соблюдается на протяжении, как минимум, пяти лет. Что касается возраста более взрослого, то здесь в значительной степени снижается подверженность нервной системы к воздействию, оказываемому фенилаланином, а также воздействию, оказываемому продуктами его распада. При соблюдении требуемых мер к 12-14 годам ребенок сможет свободно перейти к обычному питанию.
Примечательно, что медикаментозное лечение при данном заболевании носит синдромный характер. В него включено применение препаратов, ориентированных на устранение судорог, в том числе и препаратов, которые оказывают стимулирующее воздействие на интеллектуальную деятельность. В обязательном порядке детям назначается курс лечебной физкультуры и массажа. Дополнительно назначаются уроки, способствующие развитию логики.
Диагностирование фенилкетонурии производится педиатром и генетиком на основании специальных анализов в комплексе с общей характерной симптоматикой.
Все ли корректно в статье с медицинской точки зрения?
Ответьте только в том случае, если у вас есть подтвержденные медицинские знания
Заболевания со схожими симптомами:
Отек головного мозга – опасное состояние, характеризующееся чрезмерным скоплением экссудата в тканях органа. Как следствие, постепенно увеличивается его объем и растёт внутричерепное давление. Все это приводит к нарушению обращения крови в органе и к отмиранию его клеток.
Переутомление – состояние, с которым сегодня часто сталкиваются не только взрослые, но и дети. Оно характеризуется пониженной активностью, сонливостью, нарушением внимания и раздражительностью. Причём многие люди считают, что переутомление – несерьёзная проблема, и что достаточно хорошо отоспаться, чтобы оно прошло. На самом же деле избавиться от такого нарушения невозможно длительным сном. Все наоборот - постоянное желание спать и неспособность восстановить силы после сна – это основные симптомы переутомления.
Аденома паращитовидной железы представляет собой небольшое доброкачественное образование размером от 1 до 5 см, которое может самостоятельно синтезировать паратгормон, вызывая у человека симптомы гиперкальциемии. Паращитовидные железы располагаются на задней поверхности щитовидки, и их основное назначение заключается в продуцировании паратиреоидного гормона, принимающего участие в кальциево-фосфорном обмене в организме. Аденома приводит к тому, что паратиреоидного гормона начинает вырабатываться больше, чем необходимо, что и обуславливает симптомы этого заболевания.
Гидроцефалия, которую также принято определять в качестве водянки головного мозга, представляет собой заболевание, при котором происходит увеличение объема желудочков в головном мозге, причем зачастую – до весьма внушительных размеров. Гидроцефалия, симптомы которой проявляются по причине избыточной выработки ликвора (спинномозговой жидкости между сообщающимися желудочками мозга) и его скопления в области полостей головного мозга, преимущественно возникает у новорожденных, однако имеет это заболевание и место в заболеваемости других возрастных категорий.
Гидроцефалия (син. водянка) головного мозга у детей – это болезнь, характеризующаяся тем, что во внутренних его полостях и под мозговыми оболочками собирается чрезмерное количество ликвора, что также носит название спинномозговая жидкость. Причин формирования недуга достаточно много, причём они могут отличаться в зависимости от того, в каком возрасте сформировалась патология. Наиболее часто в качестве провоцирующих факторов выступают инфекционные и онкологические процессы, врождённые пороки развития и родовые травмы.
Природа не только мудра, но и щедра. Она одаривает людей многими радостями, исполняя их заветные желания иметь здоровых детей, хотя время от времени преподносит «сюрпризы», застающие молодые семьи врасплох. Один из них принадлежит к сфере генетических закономерностей. Без наследственности и изменчивости, благодаря которым у потомства появляются новые признаки, невозможна была бы эволюция жизни на Земле, однако при этом человечество отягощается грузом разнообразных мутаций, то есть изменений в наследственных структурах. Болезни, передающиеся потомству по наследству, являются частью общей наследственной изменчивости человека. Они выявляются, у новорожденных во внешне вполне благополучных семьях. В этих случаях практически здоровые родители имеют патологический задаток того или иного наследственного заболевания, о чем они не знают.
Наталья Копылова
Врач-психиатр высшей категории, к.м.н. Республиканский центр неонатального скрининга
Редкое заболевание фенилкетонурия (ФКУ) - одна из форм наследственных дефектов обмена аминокислот. В нашей стране частота этого заболевания невелика: один больной ребенок приходится на семь тысяч здоровых новорожденных. Малыш рождается с генетическим дефектом, из-за которого аминокислота фенилаланин, поступающая в организм с пищевым белком, не может превращаться в тирозин, как это бывает в норме. В результате фенилаланин и его производные накапливаются в тканях и органах малыша, оказывая токсическое воздействие на нервную систему. Если не диагностировать ФКУ в период новорожденности и, соответственно, ничего не предпринимать, болезнь приведет к весьма серьезным последствиям: у ребенка разовьется выраженная умственная отсталость - олигофрения, вплоть до крайней степени - идиотии. При отсутствии своевременного лечения больные на всю жизнь останутся глубокими инвалидами, так как повернуть болезнь «вспять» невозможно. Однако недаром говорится, что береженого Бог бережет. Поэтому родители каждого новорожденного должны понимать, как важно вовремя обследовать ребенка, чтобы не упустить время начала лечения в случае установления диагноза ФКУ.
Причины заболевания
Больной ребенок с ФКУ может родиться только в той семье, где родители практически здоровы, но оба являются носителями патологического задатка, в данном случае - носителями гена ФКУ.
Определить носительство гена ФКУ у родительской пары возможно при генетическом обследовании на гетерозиготное носительство, проводимое в некоторых федеральных медико-генетических центрах страны. Если в семье уже есть больной ребенок с ФКУ, носительство гена ФКУ у родителей очевидно.
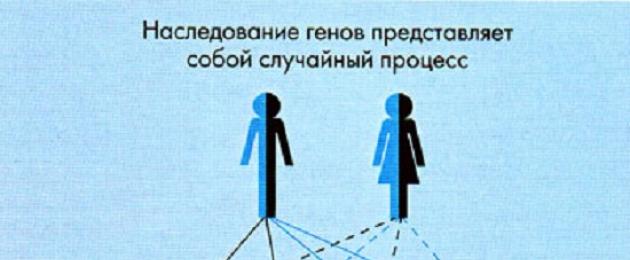
Однако даже если оба родителя являются носителями гена ФКУ, их дети не обязательно будут больны. Если принять за 100% всех детей, которые гипотетически могут родиться в данной семье, можно говорить о следующем риске возникновения заболевания ФКУ:
- риск рождения больных детей с ФКУ составляет 25%;
- риск рождения детей, являющихся, подобно их родителям, носителями гена ФКУ составляет 50%;
- в остальных 25% случаев родятся здоровые дети (см. рис.).
Известно, что носительство мутантного гена ФКУ среди населения составляет 2 -3%. Однако, как было сказано выше, риск рождения ребенка с ФКУ появится только в том случае, если оба родителя являются носителями патологического гена. Поэтому данное заболевание встречается довольно редко.
Симптомы
Ребенок с ФКУ рождается без каких-либо проявлений заболевания. Однако с началом кормления, при поступлении в организм белка грудного молока или его заменителей возникают первые микросимптомы, трудно распознаваемые не только родителями, но и педиатрами.
Так, в периоде новорожденности до начала лечения у ребенка с ФКУ возможны необоснованная вялость или беспокойство; обращают на себя внимание рассеянный, блуждающий взгляд, отсутствие улыбки, слабое двигательное оживление. К 6 месяцам у него выявляется задержка психомоторного развития: он перестает активно реагировать на происходящее; утрачивает способность узнавать мать; не переворачивается на живот; не пытается сесть.
Во втором полугодии жизни родители уже не могут не заметить непонимание речи взрослого, неумение выражать голосом и мимикой ребенка свои переживания. У детей старше трех лет нарастают умственная отсталость, возбудимость, повышенная утомляемость; нарушается поведение, что проявляется в расторможенности, психотических расстройствах.
Часто у нелеченых больных ФКУ моча имеет своеобразный «мышиный» запах. Иногда возникают судорожные приступы различной степени выраженности; экзематозные изменения на коже.
Диагностика
С первых дней после появления на свет здоровый ребенок должен очень быстро набирать темп развития, приобретая разнообразные физиологические навыки. Следите за его психомоторным развитием, постоянно консультируйтесь у врача-педиатра.
Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия - самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии.
К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних 10 лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Минздрава РФ №316 от 30.12.1993 г. организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории России. На службе у программы скрининга находятся все родовспомогательные учреждения страны, детские районные поликлиники, более 80 медико-генетических центров, консультаций и кабинетов.
Для раннего выявления ФКУ всем новорожденным проводится скрининг-тест (анализ крови). Это самый надежный способ позаботиться о здоровье малыша с первых дней его рождения.
Скрининг-тест заключается в следующем:
- У новорожденного не позднее чем на четвертый-пятый день его жизни натощак (через 3 часа после кормления) берется несколько капель крови из пяточки.
- Кровь наносится на специальный бумажный бланк, выданный лабораторией скрининга в медико-генетическом центре региона по месту рождения ребенка. Необходимо, чтобы капли крови были нанесены на бланк тремя насквозь пропитанными кровью кружками диаметром не менее 12 мм.
- Бланк с кровью как можно быстрее должен быть возвращен в лабораторию скрининга для проведения анализа крови на содержание в ней аминокислоты - фенилаланина (ФА).
- Анализ крови на ФА выполняется через сутки после поступления бланка в лабораторию.
- Результат скрининг-теста заносится в обменную карту ребенка в роддоме в виде штампа: «На ФКУ и ВГ 1 обследован».
При выписке из роддома обязательно проверьте, был ли проведен скрининг-тест на ФКУ.
Если роды происходили вне родильного дома (в обычной больнице, дома и т.д.) и скрининг-тест не производился, родителям самостоятельно без промедления следует обратиться в региональную медико-генетическую консультацию.
В Москве скрининг-тест и консультация врача-генетика проводятся в Московском центре неонатального скрининга по адресу: 119334, 5-й Донской проезд, д.21 а. Телефоны: 952-22-28, 954-41-27.
Если скрининг-тест окажется положительным, вас оповестят и пригласят на консультацию к врачу-генетику. При повторном положительном результате анализа на ФА следует немедленно начать лечение ребенка.
Диагноз ФКУ должен быть поставлен (или отвергнут) не позднее трехнедельного возраста ребенка!
Родителям не следует поддаваться панике: вероятность того, что заболевание будет обнаружено именно у вашего ребенка, очень невелика, но даже если это случится, - не волнуйтесь: развитие болезни можно предотвратить при своевременно начатом лечении.
Лечение
Единственным эффективным методом лечения больных ФКУ является специализированная диетотерапия с момента установления диагноза. Смысл диетического лечения сводится к резкому ограничению поступающего с пищей белка животного происхождения и замене его специализированными лечебными продуктами. Лечебный продукт - это сухая смесь аминокислот без фенилаланина, являющаяся практически единственным источником пищевого белка в рационе, необходимым для роста и развития ребенка. Родители детей, больных ФКУ, получают лечебные продукты в медико-генетической консультации бесплатно.
В случае выявления заболевания фенилкетонурии у новорожденного родители немедленно получают квалифицированную консультативную помощь и специализированную литературу у врача-генетика в медико-генетическом центре, консультации или кабинете по месту жительства. Госпитализации ребенка не требуется.
Течение беременности у женщины, являющейся носительницей гена ФКУ ничем не отличается от течения беременности здоровой женщины.
Желаю вам благополучных родов и исполнения заветной мечты - рождения здорового ребенка.
Фенилкетонурия – тяжелое наследственное заболевание, природа которого заключена в нарушении обмена аминокислот. Опасна тем, что тотально поражает ЦНС в результате дефицита фермента – фенилаланина, который отвечает за формирование умственной деятельности, и как следствие происходит нарушение умственного развития человека.
Причины данного заболевания вызваны генетическими факторами, поэтому фенилкетонурия наследуется. Но в то же время, родители, могут быть вполне здоровыми и не иметь проблем с обменом веществ.
Фенилаланин обеспечивает построение белка и способствует образованию гормона щитовидной железы - тироксина. И если он правильно не перерабатывается, у человека развивается слабоумие.
Спасти ребенка, который родился с фенилкетонурией возможно только при максимально раннем установлении диагноза и начале лечения. Раннюю диагностику можно провести при помощи неонатального скрининга.
Признаки
Заболевание может проявляться не сразу. Фенилаланин, который поступает с молоком матери в организм больного ребенка, а затем и с другой пищей, достигает критической концентрации и воздействует на головной мозг ребенка, нарушая, таким образом, его функционирование. Как правило, при отсутствии своевременного лечения, первые симптомы станут заметны в первые месяцы жизни ребенка.
К признакам фенилкетонурии у детей можно отнести:
- гипотонус мышц, вялость;
- снижение координации движений;
- судороги конечностей и непроизвольные движения;
- обильное срыгивание, рвоту.
Дети больные фенилкетонурией имеют схожий фенотип. Такие малыши очень миловидны, имеют белокурые волосы, голубые глаза, нежнейшую, почти прозрачную кожу. А когда болезнь начинает прогрессировать, они теряют эту ангельскую миловидность, так как в организме происходят тотальные нарушения. Телосложение больных становится непропорциональным, на коже появляются экземами, дерматиты, а лицо начинает напоминать идиопатическую маску.
Высокий процент риска у детей, оба родителя которых, являются носителями данного гена. В такой ситуации, фенилкетонурия наследуется с вероятностью 25%. А в случае, когда больной ген передается плоду лишь от одного родителя, он в 50% сам становится его носителем, не имея внешних проявлений патологии.
Болезнь может быть вызвана таким факторами как близкородственные браки, мутация гена в локализации 12 хромосомы, наследование генов ФКУ.
Симптомы
Характерно, что данное заболевание никак не проявляет себя при внутриутробном развитии плода. Ребенок рождается в срок, имеет нормальный вес, а первые 2 – 2,5 месяца после рождения ничем не отличается от здоровых сверстников.
Первым и настораживающим симптомом фенилкетонурии является частая рвота.
Больной ребенок плохо набирает в весе, не может удержать головку, не проявляет интереса к окружающему миру, а также становится сонливым и раздражительным. Такие проявления родители могут заметить в возрасте от 2 до 6 месяцев.
Наиболее характерный симптом фенилкетонурии - повышенная потливость ребенка, которая имеет крайне специфический мышиный запах. Также специалисты отмечают явное уменьшение черепной коробки, а также судороги.
К дополнительным признакам у детей можно отнести позднее прорезывание зубов, задержку двигательной активности и беспричинную агрессию.
Если на этом этапе не начнется лечение, то симптомы фенилкетонурии начнут проявляться все стремительнее и будут выражаться в диспропорциях тела, постоянном треморе конечностей, специфических позах. После 6 месяцев, такие дети уже весьма отстают в психическом развитии от своих сверстников.
Кроме указанных симптомов болезни можно указать еще синюшность конечностей, потливость, дермографизм (изменение в окраске при местном воздействии на нее). Часто у больных отмечаются, помимо указанных выше симптомов, наличие дерматита, артериальной гипотонии, тремора конечностей, частых запоров, нарушения координации движений.
Диагностика
Поскольку фенилкетонурия наследуется довольно часто, несмотря на то, что оба родители здоровы, ранняя диагностика данного заболевания является крайне важной. Она позволит избежать его прогрессирования, тяжелого развития, а также необратимых последствий.
Поэтому в роддомах на 4-5 день жизни доношенного ребенка, у него берется кровь для анализа. А у недоношенных детей – на 7 день. Данная процедура имеет название скрининг-тест крови на фенилкетонурию.
Эта процедура несложная, и предусматривает взятие капиллярной крови по прошествии часа после кормления. Данной кровью пропитывают специальный бланк, и если, концентрация превышает 2,2% фенилаланина в крови ребенка, его направляют на осмотр в медико-генетический центр.
Лечение
Единственным методом лечения фенилкетонурии является своевременное налаживание диетического питания, которое необходимо с первых дней. Его суть заключается в ограничении фенилаланина, который содержится в некоторых продуктах питания. Поэтому необходимо строго исключить все белковые продукты. Учитывая биологические процессы в организме, полное исключение фенилаланина из рациона может привести к истощению организма. Поэтому такой больной должен в обязательном порядке получать определенные аминокислотные смеси и белковые гидролизаты и соблюдать диету как минимум 5 лет.
Когда в крови больного снижается концентрация фенилаланина, постепенно в рацион вводят продукты животного происхождения. Также можно добавлять разнообразные фрукты и сезонные овощи, животные и растительные жиры, а также углеводы. При фенилкетонурии лечение предполагает постоянный контроль содержания фенилаланина в крови. Организму малыша необходимо быть снабженным этой аминокислотой в необходимом объеме для его нормального развития и роста.
Лечение заболевания медицинскими препаратами направлено на устранении симптомов. Например, при фенилкетонурии прописываются лекарства, ориентированные на устранение судорог, а также такие, которые стимулируют интеллектуальную деятельность. Необходимым является назначение курса массажа и лечебной физкультуры. Также могут предлагаться уроки на развитие логики.
Поскольку фенилкетонурия наследуется, то данное заболевание находится в компетенции врача генетика. Поэтому, его диагностирование осуществляется генетиком и педиатром на основании необходимых исследований в комплексе с общей симптоматикой. А в лечении основную роль играет соблюдение правил питания.
Согласно статистике, фенилкетонурией заболевает один ребенок из 10 тысяч новорожденных. С целью профилактики, будущим родителям стоит пройти медико-генетическое консультирование. Как правило, в группе риска пребывают те семьи, у которых были родственники, страдающие от данного заболевания.
Внимание!
Данная статья размещена исключительно в познавательных целях и не является научным материалом или профессиональным медицинским советом.
Записаться на прием к врачу
Фенилкетонурия (ФКУ, фенилпировиноградная олигофрения) — наследственное заболевание обмена веществ, обусловленное дефицитом одного из ферментов обмена фенилаланина, сопровождающееся нарушением гидроксилирования аминокислоты фенилаланина в тирозин. В результате в организме больного ребёнка происходит постепенное накопление фенилаланина и его метаболитов, оказывающих токсическое действие на ЦНС с дальнейшей задержкой психического развития. Болезнь впервые описана в 1934 г. А. Феллингом.
Код по международной классификации болезней МКБ-10:
Частота . Выявлены значительные этнические и географические различия в частоте разных мутаций при наиболее распространённой классической ФКУ. Частота классической ФКУсоставляет 1 на 4500 в Ирландии, 1 на 6000-10 000 в России, 1 на 16 000-20 000 среди белого населения США, 1 на 12 000 в Италии, 1 на 16 000 в Швейцарии и значительно снижена у афроамериканцев (1 на 50 000), китайцев и японцев, евреев ашкенази. Необычно редко наблюдают ФКУ в Финляндии (реже 1 на 100 000), к 1995 г. было выявлено четверо больных, среди полинезийцев ФКУ не выявлена. В Татарстане частота классической формы ФКУ составляет 1 на 6000 новорождённых (вне зависимости от этнической принадлежности).
Причины
Генетические аспекты . ФКУ развивается при дефектах генов следующих ферментов. ГТФ циклогидролаза 1 (233910, GCH1, 600225, 14q22.1-q22.2) . Фенилаланин гидроксилаза (261600, PAH, PKU1, 12q24.1) . Дигидробиоптеридин редуктаза (261630, QDPR, DHPR, 4p15.31) . Дигидробиоптерин синтетаза (261640, PTS, 11q22.3-q23.3).
Патоморфология — обнаружены изменения в структуре миелиновых волокон у нелеченых больных.
Симптомы (признаки)
Клиническая картина Дети с ФКУ рождаются без клинических признаков болезни. Однако фенилаланин, поступающий с первых дней жизни с молоком матери или обычной молочной смесью, способствует проявлению заболевания. Неврологические и психические расстройства.. Умственная отсталость (олигофрения, идиотия или имбецильность, глубокая психическая инвалидность). При отсутствии лечения коэффициент интеллектуального развития за каждые 10 нед снижается на 5 пунктов) .. Повышенная возбудимость в детстве.. Специфическая походка.. Специфические осанка и поза при сидении.. Необычное положение конечностей (поза портного) .. Стереотипные движения.. Повышение сухожильных рефлексов.. Судороги.. Дефектное формирование миелина. Раннее закрытие большого родничка. Микроцефалия. Изменения кожи.. Гипопигментация.. Сухость.. Экзема.. Дерматит.. Склеродермия. Волосы гипопигментированы. Рвота в периоде новорождённости. Светлые радужки, катаракта. Специфический «мышиный» запах тела и мочи.
Диагностика
Методы исследования .
Рентгенологическое исследование: мозговые кальцификаты.
Лабораторные исследования.. Дефицит гидроксилазы фенилаланина (ФКУ - 1), дигидроптеридинредуктазы (ФКУ - 2) или дигидробиоптерин синтетазы (ФКУ - 3) .. Гиперфенилаланинемия. Нормальная концентрация фенилаланина крови 58 ± 15 мкмоль/л — у взрослых, 60 ± 13 мкмоль/л — у подростков, 62 ± 18 мкмоль/л — у детей. У новорождённых верхний предел нормы составляет 120 мкмоль/л (2 мг/100 мл) .. При нелеченой классической ФКУ содержание фенилаланина крови повышается до 2,4 ммоль/л.. Фенилпировиноградная ацидемия.. Повышение в моче содержания o - гидроксифенилуксусной, фенилпировиноградной и фенилуксусной кислот и фенилацетилглютамина.
Специальные исследования: метод массового неонатального скрининга — обязательное определение содержания фенилаланина в образцах крови у всех новорождённых, полученных на 4-5 - й день жизни в родильном доме.
Молекулярная генетика: известно более 200 различных мутаций гена фенилаланингидроксилазы. Большинство из них сцеплено с определёнными гаплотипами полиморфизма длины рестрикционных фрагментов (RFLP) и числа танедемных повторов (VNTR). Один гаплотип может быть ассоциирован более чем с одной мутацией, следовательно, на основании исследования гаплотипа нельзя сделать определённого вывода о локализации мутации. Главная мутация для славянских народов — R408W/HP2/VNTR3. Исследование, проведённое в Татарстане, показало существенное различие в частоте этой мутации среди больных ФКУ русской (78%) и татарской (37%) национальностей. Интересно, что среди больных ФКУ татарской национальности часто (40%) отмечают мутации, характерные для средиземноморских, в т.ч. тюркских популяций (R261Q и др.), и не выявлено мутаций, характерных для восточных народов.
Лечение
ЛЕЧЕНИЕ
Режим амбулаторный, госпитализация показана для коррекции диеты в случае нестабильной концентрации фенилаланина плазмы.
Диета с резким ограничением содержания фенилаланина вводится с момента подтверждения диагноза классической ФКУ. Учитывая высокое содержание фенилаланина в белке, полностью исключают продукты животного происхождения (мясо, птицу, рыбу, яйца, грибы, молоко и продукты, приготовленные из них, колбасы, хлебобулочные изделия, крупы, бобовые, орехи, шоколад и др.), содержание растительных белков тщательно нормируют с учётом массы тела и возраста ребёнка. Дефицит пищевых белков и микроэлементов, возникающий вследствие длительного применения ограничительной диеты, компенсируют назначением специальных продуктов питания — смесей аминокислот или белковых гидролизатов с низким содержанием фенилаланина. Безфенилаланиновые препараты вводят с фруктовыми и овощными соками, пюре, супами. Учитывая высокую стоимость этих препаратов, лечение проводят за счёт государства под наблюдением врачей специализированного центра. При атипичных формах ФКУ (2 и 3), а также при несвоевременном ограничении продуктов в пищевом рационе даже строгое соблюдение диеты с низким содержанием фенилаланина не приводит к предупреждению тяжёлых неврологических нарушений. Назначение диеты, обогащённой тетрагидробиоптерином, также не приводит к клиническому улучшению у таких больных.
Лекарственная терапия .
Препараты выбора.. Ноотропные препараты, например пирацетам.. Иглорефлексотерапия.. Для компенсации белковой и витаминной недостаточности при назначении ограничительной диеты используют белковые гидролизаты и аминокислотные смеси, обогащённые микроэлементами и витаминами: «Лофеналак», «Фенил - 40», «Фенил - 100», Афенилак, «Аналог SP», «Эро - Бэби» (для детей до года), «Максамайд», «Максамум», «П - АМ универсальный», «ФенилФри», «Тетрафен», «Фенил - 400» (для детей после года), а также такие диетические продукты, как «PKU - 1mix», «PKU - 1» «PKU - 2» (Германия). Дозировки рассчитывают с учётом содержания естественного белка в рационе, массы тела и возраста ребёнка.. В резистентных к лечению случаях отмечают некоторый эффект от применения препаратов леводопы.
Генотерапия . Из трёх основных мероприятий, требуемых для генотерапии при фенилкетонурии, два выполнены: получена кДНК, обеспечивающая экспрессию гидроксилазы фенилаланина человека, разработана гидроксилазадефицитная животная модель. Однако векторы для эффективной передачи гена in vivo требуют дальнейшей разработки. Ретровирусные векторы, несмотря на то, что достаточно эффективны in vitro, имеют низкую эффективность передачи in vivo. Рекомбинантные аденовирусные векторы, хотя и полностью успешны на короткое время, не сохраняются в организме больше нескольких недель из - за иммунного ответа.
Течение и прогноз . Своевременно начатое диетическое лечение позволяет избежать развития клинических проявлений классической ФКУ. Необходимо проводить лечение до полового созревания, а по индивидуальным показаниям и дольше. Т.к. женщина, больная ФКУ, не может выносить здоровый плод, показано проведение специального лечения, начатого перед зачатием и продолжающегося до момента родов с целью исключить поражение плода фенилаланином плазмы крови матери.
Беременность . Повышенное содержание фенилаланина в плазме крови матери приводит к разнообразным врождённым заболеваниям плода, спектр зависит от выраженности и продолжительности повышения содержание фенилаланина. ВПС при высокой концентрации фенилаланина. Аномалии развития головного мозга, внутриутробная и послеродовая задержка роста, изменения внешности (широкая переносица с вывернутыми ноздрями) при средней концентрации фенилаланина в I триместре беременности. Неврологические симптомы при среднем повышении концентрации фенилаланина в течение всей беременности. Женщинам с классической ФКУ рекомендуют диету с низким содержанием фенилаланина, чтобы достичь концентрации фенилаланина плазмы менее 360 мкмоль/л ещё до зачатия и поддерживать эту концентрацию в течение всей беременности.
Сокращение . ФКУ — фенилкетонурия.
МКБ-10 . E70 .0 Классическая фенилкетонурия
Эта болезнь характеризуется дефицитом печеночного фермента фенилаланин гидроксилазы (другое название которого фенилаланин-4-монооксигеназы). Этот фермент катализирует превращение аминокислоты фенилаланина ("Phe) на тирозин. При дефиците фенилаланин гидроксилазы, фенилаланин не расщепляется, а накапливается и превращается в фенилпировиноградную кислоту, которая, при этом заболевании обнаруживается в моче.
С момента первого описания заболевания появилось много новых методов лечения и сегодня болезнь можно контролировать практически без возникновения каких либо побочных эффектов и неудобств, связанных с лечением. Однако, если расстройство не лечить , то его прогрессирования может привести к возникновению различных проблем, особенно с нервной системой и мозгом в частности, что в свою очередь ведет к умственной отсталости, повреждению головного мозга и вызывает появление эпилептических припадков.
Ранее, лечение ФКУ осуществлялось путем ограничения употребления фенилаланина. Однако, согласно последним исследованиям, только диетического питания может быть недостаточно, для преодоления всех негативных последствий заболевания. Оптимальное лечение заключается в снижении уровня фенилаланина до безопасного уровня и включает постоянный контроль за питанием, и когнитивным развитием. Снижение уровня фенилаланина может быть достигнуто за счет комбинированного применения продуктов с низким содержанием фенилаланина и белковых добавок. На современном этапе нет никаких эффективных лекарств от этого заболевания, однако существуют определенные препараты, предназначенные для устранения симптомов, но положительное влияние от их использования является индивидуальным в каждом отдельном случае.
Обычно фенилкетонурию определяют в процессе и при генетических исследованиях. Специализированные клиники для больных фенилкетонурией существуют во всем мире, именно в них обеспечивается постоянный уход за больными, контролируется уровень фенилаланина, умственное развитие пациентов и обеспечивается оптимальное питание.
История
Впервые фенилкетонурия была обнаружена норвежским врачом Иваром Асбьорн Феллингом
(Ivar Asbjørn Følling) в 1934 году, когда он заметил, что гиперфенилаланинемия (HPA) вызывает задержку психического развития. В Норвегии, фенилкетонурия, известная под названием болезнь Феллинга.
Д-р. Феллинг был одним из первых врачей, которые начал применять детальный химический анализ при изучении заболевания. Его осторожность и точность при проведении анализа мочи больных брата и сестры, привели к тому, что многие другие врачи (которые работали неподалеку от Осло), стали обращаться к нему с просьбой проанализировать состав мочи их пациентов. При проведении этих исследований, он обнаружил в моче восьми пациентов одно и то же вещество. Для анализа найденного вещества необходимым было проведение более основательного исследования и элементарного химического анализа. Проведя различные опыты, Феллинг выявил наличие реакций, характерных для бензальдегида и бензойной кислоты,
что позволило ему предположить, что исследуемое вещество содержит бензольное кольцо. Дальнейшая проверка показала, что температура плавления исследуемого вещества - такая же как у фенилпировиноградной кислоты, что указывало именно на ее наличие в моче. Так, тщательные исследования этого ученого вдохновили многих других исследователей проводить аналогичные детальные исследования при изучении других расстройств.
Скрининг, признаки и симптомы
Обычно, для выявления ФКУ используют высокоэффективную жидкостную хроматографию
(HPLC
), но в некоторых клиниках еще используют тест Гатри
(который ранее применялся в рамках национальной программы биохимического скрининга). В развитых странах исследования на ФКУ проводят детям, сразу после рождения.
Если ребенок не проходит обычной процедуры , которая обычно выполняется на 6 -14 день после рождения (с использованием образцов крови, полученных из пятки новорожденного ребенка), то первыми проявлениями заболевания
могут быть приступы судорог, альбинизм (очень светлые волосы и кожа), "заплесневелый запах" детского пота и мочи (возникает из-за наличия фенилацетата, одного из кетонов, образующихся). Для подтверждения или опровержения диагноза, в возрасте 2 недель, необходимо осуществить повторное исследование.
У новорожденных, больных фенилкетонурией, при рождении нет никаких видимых отклонений, но при отсутствии надлежащего лечения с самого начала, они не развиваются должным образом, кроме того, у них наблюдается и прогрессивное ухудшение мозговой деятельности (и, соответственно, развития).
В дальнейшем основными клиническими признаками
являются: гиперактивность, отклонения на электроэнцефалограмме (ЭЭГ), эпилептические припадки и трудности при обучении. Запах кожи, волос, пота и мочи (через накопление фенилацетату) - напоминает мышиный (затхлый) запах. Кроме того, у многих больных наблюдается гипопигментация и часто возникает экзема.
У тех детей, у которых заболевание было выявлено, а лечение было начато сразу после рождения, вероятность развития неврологических проблем, умственной отсталости или появления приступов судорог значительно ниже. Хотя иногда, такие клинические нарушения также могут возникать.
Патофизиология
Классическая ФКУ
, как правило, возникает в связи с , кодирующего фермент фенилаланин гидроксилазы (PAH). Этот фермент обеспечивает превращение аминокислоты фенилаланина в другие важные для организма соединения. Однако, фенилкетонурия может возникнуть и вследствие других мутаций, которые не связаны с геном PAH. Это является примером неалельной генетической гетерогенности.
Классическая ФКУ
Ген PAH находится на 12 (его 12q22-q24.1). Известно более 400 мутаций этого гена, которые вызывают появление различных заболеваний. Нарушение деятельности PAH является одной из основных причин для целого спектра заболеваний, включая классическую фенилкетонурию (ФКУ) и гиперфенилаланинемию (менее тяжелое заболевание, обусловленное накоплением фенилаланина).
ФКУ - это аутосомно-рецессивное генетическое расстройство. Это означает, что для того, чтобы ребенок унаследовал заболевание ей необходимо, унаследовать по одной мутированой копии гена от каждого из родителей. То есть родители, должны быть носителями этих дефектных генов. Однако, если только один из родителей является носителем, а у другого обе копии гена - нормальные, вероятно, что ребенок может родиться полностью здоровой.
Фенилкетонурия может возникать и у мышей, которые широко используются при исследованиях разнообразных препаратов для лечения ФКУ. Недавно было установлено последовательность генома макаки,при исследовании которой ученые обнаружили, что ген, кодирующий фенилаланин гидроксилазы имеет ту же последовательность, что и ген, который у людей отвечает за возникновение ФКУ.
Гиперфенилаланинемия, которая связана с дефицитом тетрагидробиоптерину
Любопытно, что еще одна форма гиперфенилаланемии, которая встречается очень редко и возникает когда, ген PAH функционирует нормально, но есть определенные недостатки в процессах биосинтеза или рециркуляции кофактора тетрагидробиоптерину (BH4). Этот кофактор необходим в организме для нормального функционирования фермента фенилаланин-4-монооксигеназы. Для лечения этого нарушения возможно использование коэнзима, который называется биоптерин.
Для того чтобы установить различия
между двумя выше описанными расстройствами необходимо определить уровень дофамина в организме. Как уже было отмечено, тетрагидробиоптерин необходим для превращения фенилаланина в тирозин, однако помимо этого он играет очень важную роль при преобразовании тирозина в дигидроксифенилаланин (сокращение допа, который катализируется ферментом тирозин гидроскилазой), которий в свою очередь является предшественником дофамина. Если в организме низкий уровень дофамина, то уровень пролактина - растет. Именно такой процесс характерен для гиперфенилаланинемия, связанной с дефицитом тетрагидробиоптерину, тогда как при классической ФКУ, уровень пролактина остается нормальным. Дефицит тетрагидробиоптерину может быть вызван мутациями в четырех разных генах. Согласно названия которых, именуются типы заболевания. Это:
HPABH4A, HPABH4B, HPABH4C и HPABH4D.
Метаболический путь
Обычно, фермент фенилаланин гидроксилаза участвует в превращении аминокислоты фенилаланина в аминокислоту тирозин. Если этого преобразования не происходит, то фенилаланин накапливается в организме и, соответственно, возникает дефицит тирозина. Чрезмерное количество фенилаланина может быстро разлагаться на фенил-кетоны в процессе трансаминирования глутамата. Метаболитами,
которые образуются при этой реакции, являются: фенилуксусной кислота, фенилпировиноградная кислота и фенилэтиламин. Именно поэтому для правильной диагностики фенилкетонурии, необходимо определить уровень фенилаланина в крови, если же он повышен, а в моче имеющиеся фенилкетоны, то диагноз ясен.
Фенилаланин - это большая, нейтральная аминокислота (LNAA). Эти аминокислоты «конкурируют» между собой за транспортировку через гематоэнцефалический барьер (ГЭБ) с помощью системы активного транспорта больших нейтральных аминокислот (нейтрального транспортера) (large neutral amino acid transporter, LNAAT). Повышенный уровень фенилаланина в крови, соответственно, увеличивает количество его в транспортере. Что, в свою очередь, приводит к тому, что уровень других нейтральных крупных аминокислот в мозге - снижается. Но, как известно, все эти аминокислоты необходимы для синтеза белков и нейротрансмиттеров (нейромедиаторов), именно поэтому, накопление фенилаланина нарушает процесс развития мозга, вызывая умственную отсталость.
Лечение
Если ФКУ обнаружили у ребенка сразу после рождения, то это лицо, может расти и развиваться вполне нормально, однако это возможно только в случае постоянного контроля за уровнем фенилаланина (Phe) и поддержанием его в пределах допустимых норм. Этот процесс осуществляется с помощью особой диеты, или же путем сочетания диетического питания и употребления медицинских препаратов. Как было сказано выше, когда фенилаланин не усваивается нормально организмом, то его накопление в крови токсично для мозга. Если ФКУ не лечить, то заболевание может вызвать появление следующих осложнений: тяжелую умственную отсталость, нарушение функций мозга, микроцефалию, частые смены настроения, нарушение функций моторно-двигательного аппарата и такие неврологические расстройства поведения, как СДВГ (синдром дефицита внимания и гиперактивности
).
Все больные ФКУ должны придерживаться специальной диеты, при которой ограничивается употребление фенилаланина, по крайней мере первые 16 лет их жизни. Из рациона следует исключить (или ограничить употребление) продуктов, которые содержат высокий уровень фенилаланина, в основном это: мясо, курица, рыба, яйца, орехи, сыр, бобовые, коровье молоко и другие молочные продукты. Употребление продуктов с высоким уровнем крахмала, таких как картофель, хлеб, макаронные изделия, кукуруза, тоже должно контролироваться. Маленькие дети могут продолжать употреблять грудное молоко (для получения всех полезных веществ и преимущества от грудного питания), однако его количество нужно контролировать и, кроме того, ребенку необходимо обеспечить поступление в организм всех отсутствующих питательных веществ, с помощью разнообразных пищевых добавок. Стоит отметить, что употребление многих диетических продуктов питания и диетических напитков, содержащих подсластитель аспартам, тоже следует избегать, ведь аспартам состоит из двух аминокислот: фенилаланина и аспарагиновой кислоты.
Новорожденным детям
следует вводить в рацион специальные пищевые добавки
, которые позволяют обеспечить организм необходимыми аминокислотами и другими питательными веществами, которые при низко-фенилаланиновой диете не попадают в организм с продуктами питания.
Когда ребенок подрастает, то эти специальные добавки могут быть заменены таблетками и специально разработанным рационом, в котором будут учитываться все особенности больного.
Поскольку фенилаланин необходим для синтеза многих белков, то он, бесспорно, нужен в организме, для того, чтобы обеспечить полноценное развитие человека, однако его уровень (у больных ФКУ) необходимо тщательно контролировать. Особое внимание следует обратить на добавки, содержащие тирозин, ведь именно эта аминокислота является производной от фенилаланина.
Пероральное применение тетрагидроптерину (или BH4) (который является кофактором для окисления фенилаланина) у некоторых пациентов может уменьшить уровень этой аминокислоты в крови. Фармацевтическая компания BioMarin Pharmaceutical выпустила препарат, действующим веществом в котором есть сапроптерин дигидрохлорид (Kuvan), который является одной из форм тетрагидроптерину.
Kuvan
- это первый препарат, который может обеспечить организм BH4 у больных фенилкетонурией (согласно подсчетам врачей - это около половины всех больных ФКУ), что приведет к снижению уровня фенилаланина до рекомендуемой границы. При сотрудничестве с диетологом, некоторые лица, больные фенилкетонурией (те, организм которых реагирует на лечение Kuvan) могут увеличить уровень различных природных белков в своем рационе. После больших клинических исследований, Kuvan был одобрен FDA для использования при лечении ФКУ. Некоторые исследователи и врачи, работающие с больными фенилкетонурией считают Kuvan безопасным и эффективным дополнением к диетическому питанию и, соответственно, полезным для пациентов с ФКУ.
На сегодня, другие методы лечения ФКУ еще исследуются, в том числе , замещение больших нейтральных аминокислот и соответствующих ферментов фенилаланина аммиак лиазою (PAL). Ранее, больным ФКУ разрешалось питаться без ограничений через 8 лет, позже, через 18 лет. Однако, сегодня большинство врачей рекомендуют больным ФКУ придерживаться диетического питания и контролировать уровень фенилаланина в организме на протяжении всей жизни.
Фенилкетонурия и материнство
Для беременных женщин, больных ФКУ, очень важно удерживать низкий уровень фенилаланина до и во время всей беременности, для того, чтобы ребенок был здоровым. И хотя, развивающийся плод может быть только носителем гена ФКУ, однако внутриутробная среда может иметь очень высокий уровень фенилаланина, который обладает способностью проникать через плаценту. Как следствие, у ребенка может развиваться врожденный порок сердца, возможна задержка развития, микроцефалия и умственная отсталость. Как правило, у больных фенилкетонурией женщин никаких осложнений во время беременности не возникает.
В большинстве стран, женщинам с ФКУ, которые планируют заводить детей, рекомендуется снизить уровень фенилаланина (как правило до 2-6 мкмоль / л), еще до беременности и контролировать его на протяжении всего периода вынашивания ребенка. Это достигается путем проведения регулярных анализов крови и соблюдения строгой диеты, и постоянным наблюдением врача-диетолога. Во многих случаях, как только печень плода начинает нормально производить PAH, уровень фенилаланина в крови матери падает, соответсвенно «необходимо» увеличить его, для удержания безопасного уровня - 2-6 мкмоль / л. Именно поэтому, ежедневное количество потребленного матерью фенилаланина может удвоиться или даже утроиться к концу беременности. Если же уровень фенилаланина в крови матери ниже 2 мкмоль / л, то иногда, у женщин могут возникать различные осложнения, связанные с дефицитом этой аминокислоты, такие как головная боль, тошнота, выпадение волос и общее недомогание. Если низкий уровень фенилаланина у больных ФКУ поддерживается в течение всей беременности, то риск родить больного ребенка не выше, чем у тех женщин, которые не больны ФКУ.
Дети с ФКУ могут питаться грудным молоко в сочетании с их специальными метаболическими добавками. Согласно результатам исследований, питание новорожденных, (больных ФКУ) только грудным молоком может изменить (облегчить) последствия дефицита нужных веществ, но только в том случае, когда во время кормления грудью мать придерживается строгой диеты, для удержания низкого уровня фенилаланина в организме. Однако для подтверждения этих данных, необходимы дополнительные исследования.
В июне 2010 года, американские ученые заявили, что они будут проводить детальное исследование для выявления мутаций генов в . Их основной задачей является изучение природы фенилкетонурии, которая на сегодня, встречается все чаще. Распространение заболевания связано также с тем, что больные ФКУ часто живут более 60 лет и, соответственно, часто рожают детей, которые тоже могут быть поражены этой болезнью, или быть ее носителями.
Распространенность
Фенилкетонурия встречается примерно у 1 человека на 15000 новорожденных. Но частота заболеваемости в разных популяций - отличается. Так, 1 больной ребенок среди 4500 новорожденных среди населения Ирландии, в Норвегии это соотношение составляет 1:13000, в Финляндии этот показатель еще ниже - менее чем одно лицо на 100.000 новорожденных. Чаще заболевание встречается в Турции , ведь им болеет каждый ребенок из 2600 рожденных. Болезнь также более распространена в Италии, Китае, и среди населения Йемена.






