При изучении характера наследования различных признаков у человека описаны все известные типы наследования и все типы доминирования. Многие признаки наследуются моногенно , т.е. определяются одним геном и наследуются в соответствии с законами Менделя. Моногенных признаков описано более тысячи. Среди них есть как аутосомные, так и сцепленные с полом. Некоторые из них приведены ниже.
Моногенные болезни встречаются у 1-2% населения земного шара. Это очень много. Частота спорадических моногенных болезней отражает частоту спонтанного мутационного процесса. Среди них большую долю составляют болезни с биохимическим дефектом. Типичным примером является фенилкетонурия .
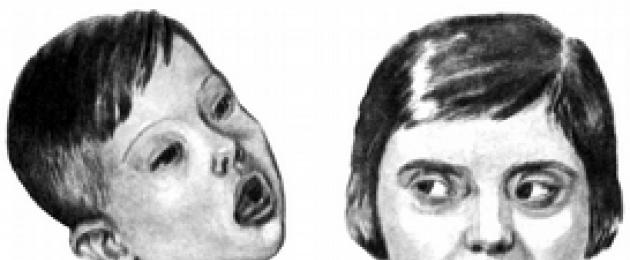
Семейное проявление
синдрома Морфана
Это тяжелое наследственное заболевание, обусловленное мутацией одного гена, нарушающей нормальный цикл превращения фенилаланина. У больных эта аминокислота накапливается в клетках. Болезнь сопровождается выраженной неврологической симптоматикой (повышенной возбудимостью), микроцефалией (маленькая голова) и в итоге приводит к идиотии. Диагноз ставится биохимически. В настоящее время в родильных домах проводится стопроцентное скринирование новорожденных на фенилкетонурию. Болезнь излечима, если вовремя перевести ребенка на специальную диету, исключающую фенилаланин.
Еще один пример моногенной болезни — синдром Морфана, или болезнь “паучьих пальцев” . Доминантная мутация одного гена имеет сильный плейотропный эффект. Помимо усиленного роста конечностей (пальцев), у больных наблюдается астения, порок сердца, вывих хрусталика глаза и другие аномалии. Болезнь протекает на фоне повышенного интеллекта, в связи с чем ее называют “болезнью великих людей”. Ею болели, в частности, американский президент А. Линкольн и выдающийся скрипач Н. Паганини.
Многие наследственные болезни связаны с изменением структуры хромосом или их нормального количества, т.е. с хромосомными или геномными мутациями. Так, тяжелое наследственное заболевание у новорожденных, известное как “синдром кошачьего крика ”, вызвано утратой (делецией) длинного плеча 5-й хромосомы. Эта мутация приводит к патологическому развитию гортани, что вызывает характерный плач ребенка. Болезнь несовместима с жизнью.

Широко известная болезнь Дауна является результатом присутствия в кариотипе лишней хромосомы из 21-й пары (трисомия по 21-й хромосоме). Причиной служит нерасхождение половых хромосом при образовании половых клеток у матери. В большинстве случаев появления у новорожденных лишней хромосомы возраст матери достигает, по крайней мере, 35 лет. Мониторинг частоты этого заболевания в районах с сильным загрязнением окружающей среды обнаружил существенное увеличение количества больных этим синдромом. Предполагается также влияние вирусной инфекции на организм матери в период созревания яйцеклетки.
Отдельную категорию наследственных болезней составляют синдромы, связанные с изменением нормального количества половых хромосом . Как и болезнь Дауна, они возникают при нарушении процесса расхождения хромосом в гаметогенезе у матери.
У человека, в отличие от дрозофилы и других животных, Y-хромосома играет большую роль в определении и развитии пола. При отсутствии ее в наборе с любым количеством Х-хромосом особь фенотипически будет женской, а ее присутствие определяет развитие в сторону мужского пола. В частности, особи мужского пола с хромосомным набором ХХY + 44А больны синдромом Клайнфельтера . Они характеризуются умственной отсталостью, непропорциональным ростом конечностей, очень маленькими семенниками, отсутствием сперматозоидов, ненормальным развитием молочных желез и другими патологическими признаками. Увеличение числа Х-хромосом в сочетании с одной Y-хромосомой не изменяет определение мужского пола, а лишь усиливает синдром Клайнфельтера. Впервые кариотип ХХYY был описан в 1962 г. у 15-летнего мальчика со значительной умственной отсталостью, евнухоидными пропорциями тела, с уменьшенными в размере яичками и оволосением по женскому типу. Подобные же признаки характерны для больных с кариотипом ХХХYY.
 Синдром Клайнфельтера (1) и синдром Тернера-Шерешевского (2)
Синдром Клайнфельтера (1) и синдром Тернера-Шерешевского (2)
Отсутствие одной из двух Х-хромосом в кариотипе женщины (ХО) вызывает развитие синдрома Тернера-Шерешевского . Больные женщины обычно низкорослы, менее 140 см, коренасты, со слабо развитыми молочными железами, имеют характерные крыловидные складки на шее. Как правило, они бесплодны из-за недоразвития половой системы. Чаще всего беременность при этом синдроме приводит к самопроизвольному аборту. Только около 2% больных женщин сохраняют беременность до конца.
Трисомия (ХХХ) или полисомия по Х-хромосоме у женщин часто вызывает заболевание, сходное с синдромом Тернера-Шерешевского.
Наследственные болезни, связанные с изменением числа Х-хромосом, диагносцируются цитологическим методом по количеству в клетках телец Барра или полового хроматина. В 1949 г. М. Барр и Ч. Бертрам, изучая интерфазные ядра нейронов у кошки, обнаружили в них интенсивно красящееся тельце. Оно присутствовало только в ядрах клеток самок. Оказалось, что оно встречается у многих животных и всегда связано с полом. Эта структура получила название полового хроматина , или тельца Барра. В ходе тщательного цитологического и цитогенетического анализа было установлено, что половой хроматин представляет собой одну из двух женских половых хромосом, находящуюся в состоянии сильной спирализации и потому неактивную. У женщин с синдромом Тернера-Шерешевского (кариотип ХО) не обнаруживается полового хроматина, так же как и у нормальных мужчин ХY. Нормальные женщины ХХ и аномальные мужчины имеют по одному тельцу Барра, а женщины ХХХ и мужчины ХХХY — по два и т.д.
Лица с наследственными заболеваниями обычно рождаются с большими физическими отклонениями, что позволяет рано диагносцировать болезнь. Но иногда заболевание не дает о себе знать месяцами и даже десятилетиями. Например, тяжелая наследственная болезнь, вызванная поражением центральной нервной системы — хорея Гентингтона — может проявиться только после 40 лет, и тогда ее носитель успевает оставить потомство. Для больных характерны непроизвольные подергивающиеся движения головы и конечностей.
Бывает так, что человек производит впечатление абсолютно здорового индивидуума, но у него есть наследственная предрасположенность к определенному заболеванию, которое проявляется под воздействием внешних или внутренних факторов. Например, некоторым людям свойственна тяжелая реакция на определенные лекарственные препараты, которая обусловлена генетическим дефектом — отсутствием в организме специфического фермента. Иногда наблюдается смертельная реакция на наркоз с виду совершенно здоровых людей, но на самом деле носящих в себе в скрытом виде особую наследственную болезнь мышц. У таких пациентов во время или после операции, проходящей под наркозом, внезапно подскакивает температура (до 42°).
Проявление у человека некоторых болезней, передаваемых по наследству, по мнению ученых связано с несколькими причинами:
- изменение числа хромосом;
- нарушения в структуре хромосом родителей;
- мутации на уровне генов.
Из общего количества всего одна пара содержит половые хромосомы, а все остальные являются аутосомными и отличаются друг от друга по размерам и форме. У здорового человека насчитывается 23 хромосомные пары. Появление лишней хромосомы либо ее исчезновение вызывают в человеческом организме различные конституционные изменения.
В результате развития современной науки ученые не только подсчитали хромосомы, но могут теперь распознать каждую пару. Проведение анализов кариотипов позволяет выявить существование наследственной болезни на ранних стадиях жизни человека. Эти изменения связаны с нарушением баланса конкретной хромосомной пары.
Причины наследственных заболеваний
Причины наследственных заболеваний , связанные с наследственными причинами, можно разделить на несколько групп:
- заболевания прямого эффекта либо врожденные; они проявляются у ребенка сразу после рождения. Типичными представителями можно назвать гемофилию, фенилкетонурию, болезнь Дауна. Возникновение подобных заболеваний ученые напрямую связывают с образом и условиями жизни, которой жили оба родителя до вступления в совместный брак и зачатия ребенка. Нередко причиной развития данного вида патологии является образ жизни будущей матери в период беременности. Чаще всего среди причин, способствующих изменениям в наборе хромосом называют употребление алкогольных напитков, наркосодержащих веществ, негативные условия экологии.
- заболевания, которые являются полученным по наследству от родителей, но активированные резким воздействием внешних раздражителей. Подобные болезни прогрессируют в процессе роста и развития ребенка, их возникновение и дальнейшее расширение спровоцирует негативность механизмов, отвечающих за наследственность. Основным фактором, запускающим в действие рост симптомов, является социально-отрицательный образ жизни. Чаще всего подобные факторы могут вызывать сахарный диабет и расстройство психики.
- заболевания, напрямую связанные с предрасположенностью, передаваемой по наследству. При наличии серьезных факторов, связанных с внешними условиями, может развиться бронхиальная астма, атеросклероз, некоторые болезни сердца, язва и прочее. Вредными факторами можно назвать некачественное питание, негативную экологию, необдуманный прием лекарств, постоянное использование средств бытовой химии.
Хромосомные наследственные изменения

Мутации, связанные с изменением числа хромосом, выглядят, как нарушение процесса деления - мейоза. В результате сбоя в «программе» происходит дубликация существующих пар хромосом, как половых, так и соматических. Зависимые от пола наследственные отклонения переносят при помощи половой Х-хромосомы.
В мужском организме эта хромосома находится без пары, тем самым заранее сохраняя у мужчин проявление наследственного заболевания. В женском организме имеется пара «Х», поэтому женщин считают носителями некачественной Х-хромосомы. Для того, чтобы хромосомные наследственные заболевания передавались исключительно по женской линии, необходимо наличие аномальной пары. Такой эффект встречается в природе достаточно редко.
Генные наследственные заболевания

Большая часть наследственных болезней происходят в результате генных мутаций, которые являются изменениями ДНК на молекулярном уровне и отлично известны ученым-генетикам и врачам-педиатрам. Различают мутации генов, которые проявляются на молекулярных, клеточных, тканевых либо органных уровнях. Не смотря на то, что промежуток от мутации на уровне молекул ДНК до основного фенотипа большой, необходимо подчеркнуть, что все возможные мутации в тканях, органах и клетках организма относятся к фенотипу. Хотя и являются сугубо внешними изменениями.
Помимо прочего, не нужно упускать из вида возможность опасного воздействия экологии и других генов, вызывающих различные модификации и реализующих функции мутирующих генов. Множественные формы белков, многообразие их функций и недостаточность научных знаний в области процессов метаболизма отрицательно влияют на попытки создания классификации генных заболеваний.
Заключение
Современная медицина насчитывает порядка 5500-6500 клинических форм генных заболеваний. Эти данные являются ориентировочными из-за отсутствия четких границ при разделении отдельных форм. Некоторые генные наследственные заболевания представляют собой различные формы с клинической точки зрения, но со стороны генетики являются последствиями мутации в одном локусе.
На начало 21 века насчитывается уже более 6 тыс. видов наследственных заболеваний. Сейчас во многих институтах мира изучаются человека, список которых огромен.
Мужское население имеет все больше генетических дефектов и все меньше шансов зачать здорового ребенка. Пока неясны все причины закономерности развития пороков, однако можно предположить, что в ближайшее 100-200 лет наука справится с решением этих вопросов.
Что такое генетические болезни? Классификация
Генетика как наука начала свой путь исследования с 1900 года. Генетическими болезнями называют те, которые связаны с отклонениями в генной структуре человека. Отклонения могут происходить как в 1 гене, так и в нескольких.
Наследственные болезни:
- Аутосомно-доминантные.
- Аутосомно-рецессивные.
- Сцепленные с полом.
- Хромосомные болезни.
Вероятность при аутосомно-доминантном отклонении - 50 %. При аутосомно-рецессивном - 25%. Болезни, сцепленные с полом, это те, что несет с собой поврежденная Х-хромосома.
Наследственнные заболевания
Приведем несколько примеров болезней, согласно вышеозвученной классификации. Итак, к доминантно-рецессивным заболеваниям относятся:
- Синдром Марфана.
- Пароксизмальная миоплегия.
- Талассемия.
- Отосклероз .
Рецессивные:
- Фенилкетонурия.
- Ихтиоз.
- Другие.
Сцепленные с полом заболевания:
- Гемофилия.
- Мышечная дистрофия.
- Болезнь Фарби.
Также на слуху хромосомные наследственные заболевания человека. Список хромосомных аномалий следующий:
- Синдром Шэрешевского-Тернера.
- Синдром Дауна.
К полигенным заболеваниям относятся:
- Вывих бедра (врожденный).
- Пороки сердца.
- Шизофрения.
- Расщепление губы и неба.
Самая распространенная генная аномалия - синдактилия. То есть срастание пальцев. Синдактилия - самое безобидное нарушение и лечится благодаря хирургии. Однако это отклонение сопровождает и другие более серьезные синдромы.
Какие заболевания наиболее опасны
Из тех перечисленных заболеваний можно выделить наиболее опасные наследственные заболевания человека. Список их складывается из тех видов аномалий, где в хромосомном наборе встречается трисомия или полисомия, то есть когда вместо пары хромосом наблюдается присутствие 3, 4, 5 или более. Встречается и 1 хромосома вместо 2. Все эти отклонения происходят из-за нарушения деления клеток.
Самые опасные наследственные заболевания человека:
- Синдром Эдвардса.
- Спинальная мышечная амиотрофия.
- Синдром Патау.
- Гемофилия.
- Другие заболевания.
Вследствие таких нарушений, ребенок проживает год или два. В некоторых случаях отклонения не столь серьезны, и ребенок может дожить до 7, 8 или даже до 14 лет.
Синдром Дауна
Синдром Дауна передается по наследству, если один или оба родителя являются носителями дефектных хромосом. Если точнее, синдром связан с хромосомы (т. е. 21 хромосомы 3, а не 2). Дети с синдромом Дауна имеют косоглазие, складку на шее, аномальную форму ушей, проблемы с сердцем и умственную отсталость. Но для жизни новорожденных опасности хромосомная аномалия не несет.

Сейчас статистика утверждает, что из 700-800 детей 1 рождается с этим синдромом. У женщин, которые хотят завести ребенка после 35, больше вероятность родить такого малыша. Вероятность где-то 1 к 375. А вот женщина, решившаяся на рождение малыша в 45, имеет вероятность 1 к 30.
Акрокраниодисфалангия
Тип наследования аномалии - аутосомно-доминантный. Причина синдрома - нарушение в 10 хромосоме. В науке эта болезнь называется акрокраниодисфалангия, если проще, то синдром Аперта. Характеризуется такими особенностями строения тела, как:
- брахикефалия (нарушения соотношения ширины и длины черепа);
- срастание коронарных швов черепа, вследствие этого наблюдается гипертензия (повышенное кровяное давление внутри черепа);
- синдактилия;
- выпуклый лоб;
- часто умственная отсталость на фоне того, что череп сдавливает мозг и не дает расти нервным клеткам.

В наше время детям, имеющим синдром Аперта, назначают хирургическую операцию по увеличению черепа, чтобы восстановить кровяное давление. А психическую недоразвитость лечат стимуляторами.
Если в семье есть ребенок, у которого диагностирован синдром, вероятность того, что 2 ребенок родится с тем же отклонением, очень велика.
Синдром счастливой куклы и болезнь Кэнэвэн — ван-Богарта — Бертрана
Рассмотрим подробнее и эти заболевания. Опознать синдром Энгельмана можно где-то с 3-7 лет. У детей бывают судороги, плохое пищеварение, проблемы с координацией движений. У большинства из них косоглазие и проблемы с мышцами лица, из-за чего на лице очень часто улыбка. Движения ребенка очень скованы. Для врачей это понятно при попытках ребенка ходить. Родители в большинстве случаев не знают, что происходит и тем более с чем это связано. Чуть позже заметно и то, что они не могут говорить, лишь стараются что-то нечленораздельно пробормотать.

Причина, по которой у ребенка проявляется синдром - это проблема в 15 хромосоме. Встречается заболевание крайне редко - 1 случай на 15 тыс. рождений.
Другая болезнь - болезнь Кэнэвэн - характеризуется тем, что ребенок имеет слабый мышечный тонус, у него проблемы с проглатыванием пищи. Заболевание обусловлено поражением центральной нервной системы. Причина - поражение одного гена в 17 хромосоме. Вследствие этого нервные клетки головного мозга разрушаются с прогрессирующей быстротой.

Признаки болезни можно увидеть в 3-хмесячном возрасте. Проявляется болезнь Кэнэвэн так:
- Макроцефалия.
- Судороги появляются в месячном возрасте.
- Ребенок не способен вертикально держать голову.
- После 3 месяцев повышаются сухожильные рефлексы.
- Многие дети к 2 годам слепнут.
Как видим, очень разнообразны наследственные заболевания человека. Список, приведенный лишь для примера, далеко не полный.
Хотелось бы отметить, что если у обоих родителей есть нарушение в 1 и том же гене, то шансы родить больного ребенка велики, но если аномалии в разных генах, то можно не опасаться. Известно, что в 60 % случаях хромосомные аномалии у зародыша приводят к выкидышу. Но все же 40 % таких детей рождаются и борются за свою жизнь.
Ученые утверждают, что внешний вид человека, состояние здоровья и прочие индивидуальные особенности зависят от двух главных факторов: и влияния окружающей среды. Причем на долю генетики приходится 70%.
Большинство заболеваний в той или иной мере связаны с наследственностью: иногда из-за генетики повышается риск развития определенной болезни, но есть и ряд недугов, непосредственно связанных с поломкой в генетическом аппарате. Однако не все потеряно: у каждого из нас есть шанс повлиять на свою судьбу, ведь 30% здоровья зависит от образа жизни, характера питания, физических нагрузок и усилий врачей.
Особенности болезней, передающихся по наследству
Врожденные и наследственные болезни - не одно и то же, хотя и те, и другие берут начало с момента рождения младенца.
Врожденные болезни формируются в результате нарушения течения беременности, влияния алкоголя, никотина, некоторых лекарств и заболеваний ( , вирусный гепатит, ). При этом плод изначально был здоров.
Болезни с наследственной предрасположенностью не оставляют ребенку даже призрачного шанса. В этом случае поломка происходит гораздо раньше - на этапе передачи генетического материала от родителей к детям.
Второй особенностью наследственных недугов является невозможность полного излечения. Пневмонию и ангину можно вылечить приемом антибиотиков, воспаленный аппендикс или желчный пузырь - удалить. А вот исправить генетический материал пока не представляется возможным. Ученые пытаются исправлять генетический материал, но до внедрения наработок в широкую практику пока далеко.
Единственным возможным способом лечения наследственных болезней является терапия, направленная на устранение симптомов и улучшение качества жизни. В ряде случаев дает эффект медикаментозная профилактика обострений, но прогноз все равно остается неутешительным. Наследственные болезни, к великому сожалению, до сих пор неизлечимы.
5 самых популярных наследственных заболеваний
Близорукость - самая частая наследственная болезнь1. Близорукость
Это, пожалуй, одна из наиболее распространенных болезней, напрямую передающихся по наследству. Конечно, неправильная поза при чтении, частый просмотр телевизора, ежедневное многочасовое сидение перед экраном ноутбука и отсутствие в рационе достаточного содержания тоже играют роль в ухудшении зрения.
Однако в одном и том же классе школы найдутся дети, которые ведут себя одинаково - при этом один уже в очках, а другой четко видит. Главная причина близорукости - отягощенная наследственность.
Причина болезни - особенность мышц, способствующих вытягиванию глазного яблока. В результате изображение фокусируется не на сетчатке глаза, а ближе, и человек видит нечетко.
Если мать или отец страдали миопией, то вероятность передачи ребенку составляет 30-40%, а если оба - то 70%. Заболевание чаще проявляется в период активного роста - в подростковом возрасте, но заболеть может уже и младший школьник.
Это классическое наследственное заболевание. Существуют несколько подвидов гемофилии, при которых поломка приводит к нарушению выработки отдельных факторов свертывания. Степень тяжести также неодинакова. Выделяют три вида заболевания: гемофилия А, В и С.
Мутация, которая привоит к гемофилии, сцеплена с X хромосомой. У женщин две X хромосомы, поэтому, если в одной из них есть эта аномалия, то дама не болеет, а просто становится носительницей. История насчитывает всего 60 случаев, когда патология затрагивала сразу две хромосомы, и женщина заболевала.
Практически все больные гемофилией - мальчики, ведь у них одна Х хромосома. Одним из самых известных гемофиликов был малолетний царевич Алексей Николаевич. Ко дню расстрела в 14 лет мальчик находился в крайне тяжелом состоянии.
3. Тромбофилия
Тромбофилия - патологическое состояние, при котором повышается свертываемость крови. Есть множество разновидностей тромбофилии, при которых мутации происходят в отдельных звеньях свертывающей системы (например, дефицит антитромбина, протеина C и S, и антифосфолипидный синдром).
Многим кажется, что это состояние редкое и их оно не коснется. И, тем не менее, именно тромбофилия часто приводит ишемическим инфарктам, инсультам, тромбоэмболиям легочной артерии, тромбозам сосудов у людей до 40 лет.
Нередко тромбофилию выявляют при обследовании по поводу привычных выкидышей и не вынашивания беременности у женщин. К сожалению, велика вероятность, что данное состояние унаследуют дети больных.
Этот недуг встречается у одного из 2500 новорожденных, что не так уж и редко. Муковисцидоз наследуется по аутосомно-рецессивному типу. То есть для того, чтобы родился больной ребенок, малыш должен получить неправильный ген от матери и отца одновременно.
От 2 до 5% людей во всем мире являются носителями муковисцидоза, и даже понятия об этом не имеют. Если они встретят такого же, как он, то могут родить больного ребенка с вероятностью 25%.
Муковисцидоз связан со снижением выработки секрета всеми железами организма. В результате нарушается работа дыхательной и пищеварительной системы. В частности, не выделяется секрет из просвета бронхов при респираторных заболеваниях, и отсутствует выработка ферментов для переваривания пищи поджелудочной железой.
Лечение заключается лишь в заместительной терапии, а прогноз остается неблагоприятным. В Европе такие люди доживают до 40 лет, в России - максимум до 28.
5. Миодистрофия
Это страшное заболевание включает в себя сразу несколько подвидов (Эрба-Рота, Ландузи, Дюшенна). Суть болезни заключается в прогрессирующей мышечной слабости, постепенно приводящей к полному обездвиживанию человека.
Однако, учитывая тот факт, что болезнь передается с рецессивным геном, ребенок с миодистрофией может родиться у внешне здоровых родителей Достаточно, чтобы вероятность носительства у родителей составила 25%.
Как правило, первые признаки миопатии Дюшенна врачи выявляют в возрасте 6 месяцев. Иногда их даже «списывают» на осложнение вакцинации АКДС, что в корне неверно, ведь болезнь наследственная. Юношеская форма Эрба-Рота дебютирует в 14-16 лет.
Лечение миодистрофии симптоматическое, и направлено лишь на улучшение качества и максимального продления жизни.
Можно ли предотвратить генетические заболевания
Ученым пока не известны способы излечения генетических заболеваний, однако такие попытки предпринимаются по всему мируПредотвратить появление наследственных болезней на сегодняшний день невозможно. Однако можно пройти обследование на наиболее распространенные виды мутаций, и выявить вероятность рождения ребенка с патологией в конкретной паре.
Дальше многое зависит от поведения родителей. Изменение образа жизни, конечно, не повлияет на генетический материал, однако в ряде случаев снижает риск тяжелых проявлений болезни.
Поэтому не стоит бояться генетических тестов: чем раньше будет поставлен диагноз, тем легче будет помочь ребенку. Если вы не знаете, в какую лабораторию обратиться, бесплатно подберет лабораторию, в которой можно сделать генетический тест на врожденные болезни по приемлемой цене.
9.1 Понятие, классификация и особенности наследственной патологии
Патология – это любое отклонение от нормального течения биологических процессов – обмена веществ, роста, развития, размножения.
Наследственная патология – отклонение от нормы с установленным фактом наследования, то есть передачи от поколения к поколению. Следует различать врожденную патологию – присутствующую от рождения индивидуума – от наследственной патологии. Врожденная патология может быть обусловлена действием факторов внешней среды – недостатком питательных веществ и кислорода во время внутриутробного развития, родовыми травмами, инфекциями и так далее. Установление в соответствие с требованиями генетического анализа (глава II) факта наследования аномального признака является единственным основанием признания наследственного характера патологии.
Существует два типа классификации наследственной патологии. Первый (принятый преимущественно в отечественной литературе) – клинический тип. Согласно этому типу классификации существует четыре группы заболеваний:
Группа I – это собственно наследственные болезни - хромосомные и генные заболевания (синдромы Эдвардса и Патау, фенилкетонурия, муковисцедоз);
Группа II – болезни с выраженной наследственной предрасположенностью, в патогенезе которых проявление наследственных факторов определяется действием специфических внешних обстоятельств (артериальная гипертензия, сахарный диабет, подагра);
Группа III – заболевания, которые определяются преимущественно факторами внешней среды, но в патогенезе которых некоторую роль играют наследственные факторы (глаукома, атеросклероз, рак молочной железы);
Группа IV – болезни, к которым наследственность на первый взгляд не имеет отношения (пищевые отравления, переломы, ожоги).
Следует отметить, что часто используемые понятия «семейные» и «спорадические» заболевания не имеют прямого отношения к наследственности. Семейные заболевания наблюдаются у родственников, но могут быть вызваны и действием одинаковых внешних причин, например, характером питания. Спорадические случаи наблюдаются у отдельных индивидуумов, но могут быть обусловлены и редким сочетанием аллелей или возникшей de novo мутацией.
Вторая система классификации – генетическая – является общепринятой в зарубежной литературе и в последнее время находит все более частое применение и в литературе на русском языке. Согласно этой системе выделяют пять групп:
Группа I – генные болезни, определяемые мутациями в определенных генах. Это преимущественно моногенные признаки с аутосомно-доминантным, аутосомно-рецессивным, сцепленным с полом доминантным, сцепленным с полом рецессивным, голандрическим и митохондриальным типом наследования (глава II);
Группа II – хромосомные болезни, то есть геномные и хромосомные мутации (глава V);
Группа III – болезни с наследственной предрасположенностью, в патогенезе которых играют роль средовые и наследственные факторы, имеющие моногенный или полигенный тип наследования (миопия, патологическое ожирение, язва желудка).
Группа IV – генетические болезни соматических клеток, зачастую связанные со злокачественными новообразованиями (ретинобластома, опухоль Вильмса, некоторые формы лейкемии);
Группа V – болезни генетической несовместимости матери и плода, которые развиваются в результате иммунной реакции матери на антигены плода (несовместимость по резус-фактору и некоторым другим эритроцитарным системам антиген-антитело).
Наследственные заболевания могут начать свое проявление в разном возрасте. Характер манифестации (времени проявления первых симптомов болезни) является специфическим для разных форм наследственной патологии. Как правило, для наследственных заболеваний характерно хроническое (продолжительное) прогредиентное (с нарастанием степени выраженности симптомов) течение.
9.2 Хромосомные болезни
К этой группе относят заболевания, вызванные аномалиями числа или структуры хромосом. Около 1% новорожденных имеют аномальный кариотип, а среди мертворожденных встречаемость аберраций числа или структуры хромосом – 20%. Общими характерными чертами хромосомных болезней являются: низкий вес при рождении, задержка развития, низкий рост, микроцефалия, микрогнатия, нарушения остеогенеза, аномальное расположение глаз. Более подробное описание хромосомных болезней приведено в разделах 5.8 и 5.9.
9.3 Генные болезни
Генными болезнями называют патологические состояния, причиной которых являются генные мутации. Чаще всего это понятие применяют к моногенным заболеваниям.
Для этой группы характерна гетерогенность – одинаковые заболевания могут быть вызваны мутациями в разных генах. Общими принципами развития патологии на уровне генов могут быть:
Выработка аномального белкового продукта;
Отсутствие нормального белка;
Недостаточное количество нормального белка;
Избыток нормального белкового продукта.
По характеру нарушений гомеостаза (постоянства внутренней среды организма) выделяют следующие группы генных болезней:
1. Болезни аминокислотного обмена.
Самая многочисленная группа наследственных болезней обмена веществ. Почти все они наследуются по аутосомно-рецессивному типу. Причина заболеваний - недостаточность того или иного фермента, ответственного за синтез аминокислот.
Фенилкетонурия - нарушение превращения фенилаланина в тирозин из-за резкого снижения активности фенилаланингидроксилазы – аутосомно-рецессивное заболевание. Проявляется в возрасте 2-4 месяцев, первые симптомы – вялость, судороги, экзема, «мышиный» запах (запах кетонов). Постепенно развиваются тяжелые поражения головного мозга, приводящие к резкому снижению интеллекта вплоть до идиотии. Если с первых дней жизни полностью исключить (или существенно ограничить количество) фениаланин из рациона больного ребенка до полового созревания, симптомы не развиваются. Болезнь обусловлена мутациями в гене PAH , который кодирует фенилаланин-4-гидроксилазу. Ген PAH локализован в HSA12q24.1. Описано несколько десятков мутаций этого гена в разных популяциях. Существуют диагностические системы на основе ПЦР, которые позволяют выявлять гетерозиготное носительство. В последнее время разрабатываются новые подходы к лечению феникетонурии - заместительная терапия фениаланинлиазой – растительным ферментом, который катализирует расщепление фенилаланина на безвредные метаболиты, - и генная терапия путем встраивания в геном нормального гена фениаланингидроксилазы.
Алкаптонурия – аутосомно-рецессивное нарушение обмена тирозина и накопления в тканях организма (суставные хрящи, сухожилия) гомогентизиновой кислоты. Манифестация происходит в детском возрасте. Первый симптом – потемнение мочи. Часто развивается мочекаменная болезнь и пиелонефрит. Накопление продуктов распада гомогентизиновой кислоты приводит к поражению суставов (в первую очередь коленных и тазобедренных). Отмечается потемнение и повышенная хрупкость соединительной ткани. Характерно потемнение склер и ушных раковин. Мутации в гене HGD – оксидазы гомогентизиновой кислоты – являются причиной этого заболевания. Этот ген содержит 14 экзонов и локализован в HSA3q21-23. Описано около 100 различных миссенс-мутаций, мутаций типа сдвига рамки считывания и изменения сайта сплайсинга, которые связаны с этим заболеванием.
Глазо-кожный альбинизм 1 – отсутствие или существеный недостаток пигмента кожи, волос, радужной и пигментной оболочек глаза (Рисунок IX, 1).
Рисунок IX, 1. Представитель негроидной расы - альбинос. По материалам сайта http://upload.wikimedia.org/wikipediacommons/99a/Albinisitic_man_portrait
Заболевание с аутосомно-рецессивным типом наследования. Проявляется в различной степени депигментации кожи, волос, радужной и пигментной оболочек глаза, снижением остроты зрения, светобоязни, нистагме, частых солнечных ожогах. Различные миссенс-мутации, мутации типа сдвига рамки считывания и нонсенс мутации в гене тирозиназы (TYR , HSA11q24) ответственны за это заболевание.
2. Нарушения обмена углеводов
Галактоземия – отсутствие или существенное снижение активности фермента галактозо-1-фосфат-уридилтрансферазы и накопление в крови галактозы и ее производных, которые оказывают токсическое действие на центральную нервную систему, печень и хрусталик глаза. В первые дни и недели жизни наблюдаются желтуха, увеличение печени, нистагм, гипотония мышц, рвота. Со временем развивается катаракта, отставание в физическом и умственном развитии. Характерна непереносимость молока.
Болезнь имеет аутосомно-рецессивный тип наследования. Несколько форм этого заболевания обусловлены различными мутантными аллелями гена GALT (галактозо-1-фосфатуридилтрансферазы), локализованного в районе HSA9p13. Миссенс-мутации в разной степени снижают активность фермента, что определяет разную степень выраженности симптомов заболевания. Например, галактоземия Дурте протекает почти бессимптомно, отмечается только склонность к расстройствам печени.
Болезнь Гирке (гликогеноз I типа, гликогеновая болезнь I типа) – неспособность превращения глюкозо-6-фосфата в глюкозу, которая приводит к нарушению синтеза и разложения гликогена. Депонирование гликогена происходит, обратный процесс – нет. Развивается гипогликемия. Накопление избыточного количества гликогена в печени и почках приводит к печеночной и почечной недостаточности. Тип наследования – аутосомно-рецессивный. Причина заболевания – мутация в гене G6PC , который кодирует фермент глюкозо-6-фосфатазу. Описано 14 мутантных аллелей этого гена, которые связаны с болезнью Гирке. Существуют молекулярно-генетические тесты для выявления гетерозиготного носительства и пренатальной диагностики этого заболевания.
3. Нарушения липидного обмена
Болезнь Ниманна-Пика типов А и Б - снижение активности фермента кислой лизосомальной сфингомиелиназы, который кодируется геном SMPD1 (HSA11p15.4-p15.1). Тип наследования – аутосомно-рецессивный. Нарушение липидного метаболизма приводит к накоплению липидов в печени, легких, селезенке, нервных тканях. Характерна дегенерация нервных клеток, нарушение деятельности нервной системы, повышенный уровень холестерина и липидов в крови. Тип А летален в раннем детском возрасте. Тип Б протекает более мягко, больные как правило доживают до взрослого состояния. Разные типы обусловлены разными мутациями в гене SMPD1.
Болезнь Гоше (гликозилцерамидный липидоз) - накопление глюкоцереброзидов в клетках нервной и ретикуло-эндотелиальной системы, обусловленное дефицитом фермента глюкоцереброзидазы, которая кодируется геном GBA (HSA1q21). Относится к группе лизосомных болезней накопления. Некоторые формы заболевания проявляются в тяжелых поражениях печени, селезенки, нервной и костной тканей.
4. Наследственные болезни пуринового и пиримидинового обмена
Синдром Леша-Нихена – сцепленное с полом рецессивное заболевание, при котором резко возрастает содержание мочевой кислоты во всех жидкостях тела. Последствием этого является задержка развития, умеренная умственная отсталость, приступы агрессивного поведения с самоповреждением. Недостаточность ферментативной активности гипоксантин-гуанинфосфорибозилтрансферазы по причине мутаций в гене HPRT1 (HSAXq26-q27.2) лежит в основе этого заболевания. Описаны несколько мутаций в том же гене, следствием которых является подагра (нарушение пуринового обмена и отложение мочекислых соединений в тканях).
5. Нарушения обмена соединительной ткани
Синдром Марфана («паучьи пальцы», арахнодактилия) - поражение соединительной ткани вследствие мутации в гене FBN1 (HSA15q21.1), ответственном за синтез фибриллина. Наследуется по аутосомно-доминантному типу. Клиническая полиморфность заболевания объясняется большим числом мутантных аллелей, каждый из которых может проявляться в гетерозиготном состоянии. Для больных характерен высокий рост, астеническое телосложение (непропорционально длинные конечности), арахнодактилия (длинные тонкие пальцы), слабость связочного аппарата, отслойка сетчатки глаза, подвывих хрусталика, пролапс митрального клапана (Рисунок IX, 2).

Рисунок IX, 2. Синдром Марфана. По материалам сайта http://www.spineinfo.ru/infosources/case/cases_14.html.
Мукополисахаридозы - группа заболеваний соединительной ткани, связанных с нарушеним обмена кислых гликозаминогликанов (мукополисахаридов), вызванных недостаточностью некоторых лизосомных ферментов. Эти заболевания относят к лизосомным болезням накопления. Они проявляются в различных дефектах костной и соединительной тканей. Мукополисазаридоз типа I (синдром Хурлера) – аутосомно-рецессивное заболевание, возникающее в результате дефицита фермента альфа-L-идуронидазы из-за мутаций в гене IDUA (HSA4q16.3). Это приводит к накоплению белково-углеводных комплексов и жиров в клетках организма. В результате у больных наблюдается малый рост, существенная задержка умственного развития, увеличение печени и селезенки, пороки сердца, помутнение роговицы, деформация костей и огрубение черт лица (Рисунок IX, 3).

Рисунок IX, 3. Синдром Хурлера. По материалам сайтаhttp://medgen.genetics.utah.edu/photographs/pages/hurler_syndrome.htm.
Мукополисахаридоз типа II (синдром Хантера) – сцепленное с полом рецессивное заболевание, которое обусловлено дефектом фермента идуронатсульфотазы из-за мутации в гене IDS (HSAXq28). Веществами накопления являются дерматан- и гепарансульфаты. Характерны грубые черты лица, скафоцефалия, шумное дыхание, низкий грубый голос, частые острые респираторные вирусные инфекции (Рисунок IX, 4) . В возрасте 3-4 лет появляются нарушения координации движений - походка становится неуклюжей, дети при ходьбе часто падают. Для больных характерны эмоциональная лабильность и агрессивность. Наблюдаются также прогрессирующая тугоухость, узелковые поражения кожи спины, остеоартриты, поражения роговицы.
\ 
Рисунок IX, 4. Синдром Хантера. По материалам сайта http://1nsk.ru/news/russia/23335.html.
Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо)- заболевание, вызванное накоплением гепарансульфата. Для него характерна генетическая гетерогенность – существуют 4 типа этой болезни, вызванные мутациями в 4-х разных генах, кодирующих ферменты, участвующие в метаболизме накапливаемого вещества. Первые симптомы болезни в виде нарушений сна появляются у детей старше 3 лет. Постепенно развивается апатия, отмечается задержка психомоторного развития, нарушения речи, черты лица становятся грубыми. Со временем дети перестают узнавать окружающих. Для больных арактерны задержка роста, контрактуры суставов, гипертрихоз, умеренная гепатоспленомегалия. В отличие от синдромов Хурлера и Хантера при болезни Санфилиппо преобладает умственная отсталость, а поражения роговицы и сердечно-сосудистой системы отсутствуют.

Рисунок IX, 5. Синдром Санфилиппо. По материалам сайта http://runkle-science.wikispaces.com/Sanfilippo-syndrome.
Фибродисплазия (оссифицирующий миозит, параоссальная гетеротопическая оссификация, болезнь Мюнхеймера) - заболевание соединительной ткани, связанное с ее прогрессирующим окостенением в результате мутации в гене ACVR1 (HSA2q23-q24), который кодирует рецептор активина А. Тип наследования – аутосомно-доминантный. Заболевание проявляется врожденными дефектами развития - прежде всего искривленными большими пальцами стоп и нарушениями в шейном отделе позвоночника на уровне позвонков с2 - с7. Заболевание имеет прогредиентный характер, приводит к значительным нарушениям функционального состояния опорно-двигательного аппарата, глубокой инвалидизации больных и смерти преимущественно в детском и молодом возрасте (Рисунок IX, 6). Болезнь еще называют «болезнь второго скелета», так как там где в организме должны происходить штатные противовоспалительные процессы, начинается рост кости.

Рисунок IX, 6. Фибродисплазия. По материалам сайта http://donbass.ua/news/health/2010/02/15.
6. Нарушения циркулирующих белков
Гемоглобинопатии - наследственные нарушения синтеза гемоглобина. Различают две группы гемоглобинопатий. Для первой характерно изменение первичной структуры белка глобина, что может сопровождаться нарушениями его стабильности и функции (например, серповидноклеточная анемия ). При гемоглобинопатиях второй группы структура гемоглобина остается нормальной, снижена лишь скорость синтеза глобиновых цепей (например, β -талассемия ).
7. Нарушения обмена веществ в эритроцитах
Наследственный сфероцитоз - врождённая недостаточность липидов оболочки эритроцитов. Для заболевания характерен аутосомно-доминантный или аутосомно-рецессивный тип наследования в зависимости от мутации гена SPTA1 (HSA1q21), который кодирует эритроцитарный α-1 спектрин. Аномалия этого белка приводит к повышению концентрации ионов натрия внутри эритроцита, и проникновению в него избытка воды из-за повышения осмотического давления. Вследствие этого образуются сферические эритроциты – сфероциты, которые в отличие от двояковогнутых нормальных эритроцитов, не обладают способностью изменять форму в узких участках кровотока, например при переходе в синусы селезенки. Это приводит к замедлению продвижения эритроцитов в синусах селезенки и отщеплению части мембраны эритроцита с образованием микросфероцитов. Разрушенные эритроциты поглощаются макрофагами селезенки. Гемолиз эритроцитов приводит к гиперплазии клеток пульпы и увеличению селезенки. Одним из основных клинических симптомов является желтуха. Основными симптомами наследственного сфероцитоза являются увеличение селезенки (обычно выступает из-под подреберья на 2 - 3 см) и желтуха. Иногда наблюдаются признаки замедленного развития, нарушения лицевого скелета, башенный череп, седловидного носа, высокого стояния неба, нарушения расположения зубов, узких глазниц.
8. Наследственные болезни обмена металлов
Болезнь Коновалова-Вильсона (гепатоцеребральная дистрофия) – аутосомно-рецессивное нарушение метаболизма меди, приводящее к тяжелейшим поражениям центральной нервной системы и внутренних органов. Заболевание обусловлено низким или аномальным синтезом церулоплазмина (белка, транспортирующего медь) из-за недостаточности ферментативной активности медь-переносящей АТФазы. Мутации (их описано около 200) в гене ATP7B (HSA13q14-q21) приводят к изменениям β-полипептида этого фермента, что является генетической основой этой патологии. Основную роль в патогенезе играет нарушение обмена меди, её накопление в нервной, почечной, печёночной тканях и роговице, вследствие чего происходит токсическое повреждение медью данных органов. В печени формируется крупноузловой или смешанный цирроз. В почках в первую очередь страдают проксимальные канальцы. В головном мозге поражаются в большей степени базальные ганглии, зубчатое ядро мозжечка и черная субстанция.
9. Нарушения всасывания в пищеварительном тракте
Муковисцидоз (кистозный фиброз) - аутосомно-рецессивное заболевание, характеризующееся поражением желез внешней секреции, тяжёлыми нарушениями функций органов дыхания и желудочно-кишечного тракта. Причиной являются мутации гена CFTR (HSA7q31.2), который кодирует трансмембранный регулятор кистозного фиброза. Заболевание характеризуется поражением желез внешней секреции, тяжёлыми нарушениями функций органов дыхания и желудочно-кишечного тракта.
Непереносимость лактозы (гиполактазия) – аутосомно-рецессивное патологическое состояние плохого усвоения лактозы (молочного сахара), генетической основой которого являются мутации в регуляторной и кодирующей областях гена LCT (HSA2q21), который кодирует лактазу. Этот фермент экспрессируется преимущественно в ресничных клетках кишечника и отвечает за расщепление лактозы на галактозу и глюкозу. Основными симптомами лактазной недостаточности являются метеоризм, боли в животе, диарея, рвота. У детей лактазная недостаточность может проявляться хроническими запорами, беспокойством и плачем после еды. В разных популяциях человека частоты мутантных аллелей варьируют от 1 до 100%.
10. Гормональные нарушения
Тестикулярная феминизация (синдром Морриса) – сцепленное с полом рецессивное заболевание, когда при мужском кариотипе (46, XY) проявляется женский фенотип. Экспрессивность варьирует. При неполной феминизации гонады развиваются по мужскому типу, но некоторые половые признаки соответствуют женскому полу с разной степенью выраженности - гипертрофированный клитор, неполное закрытие шва мошонки, мошонкообразные большие половые губы, укороченное влагалище (Рисунок IX, 7). При полной феминизации основным симптомом является отсутствие менструаций и полового оволосения при хорошо развитых молочных железах и женском фенотипе. Причиной заболевания являются различные мутации в гене AR (HSAXq11-q12), который кодирует рецептор андрогена.

Рисунок IX, 7. Вид наружных половых органов при неполной тестикулярной феминизации. По материалам сайта http://www.health-ua.org/img/woman/tabl/8_17.jpg.
Андрогенитальный синдром (женский псевдогермафродитизм) – эндокринное нарушение с аутосомно-рецессивным типом наследования, при котором больная имеет наружные половые органы мужского типа и женскую гормональную структуру. У больных увеличен клитор, который становится похож на мужской половой член с одним уро-генитальным отверстием, отсутствует наружный вход во влагалище, малые половые губы отсутствуют, большие губы похожи на «разрубленную» мошонку. При этом внутренние половые органы могут иметь нормальный вид. Генетической основой заболевания являются мутации гена CYP21 (HSA6q21.3), который кодирует фермент 21-гидроксилазу группы цитохрома П450, участвующий в синтезе гормонов альдостерона и кортизола.
9.4 Молекулярные маркеры в изучении наследственной патологии
Значительная часть наследственных болезней и болезней с наследственной предрасположенностью имеют не моногенную природу. Их можно отнести к количественным признакам, то есть тем, которые имеют непрерывный ряд изменчивости и могут быть измерены – например, рост, вес, длина конечностей. Аллели большого числа генов вносят вклад в проявление таких признаков, поэтому их называют полигенными. Проследить их наследование и выявить гены, аллели которых участвуют в патологических процессах, можно при помощи генетических маркеров. Выявление сцепленного наследования (ассоциации) фенотипических признаков с генетическими маркерами позволяет найти районы хромосом, оказывающие решающее влияние на изучаемые процессы (позиционное клонирование), и получить надежные системы для молекулярной диагностики (молекулярное маркирование). В настоящее время наиболее распространенными маркерами в генетике человека являются микросателлитные локусы (рисунок IX, 8; раздел 8.1) и мононуклеотидные полиморфные сайты – SNP (рисунок IX, 9), основные особенности которых показаны в таблице IX, 1.
Анализ экспрессии генов (всех или группы) на биочипах в тканях, имеющих отношение к определенному наследственному заболеванию, в норме и патологии часто позволяет выявить гены-кандидаты для изучаемой болезни. Хромосомную локализацию последовательностей ДНК, влияющих на количественный признак (QTL), можно определить на основе совместного наследования с несколькими близко расположенными маркерами. Если удается найти маркеры, ограничивающие QTL с двух сторон, то на основе данных геномного сиквенса (разделы 7.7 и 8.4) можно составить список генов, являющихся позиционными кандидатами для QTL изучаемого заболевания. При одновременном использовании анализа экспрессии и исследования ассоциаций заболевания с молекулярными маркерами можно определить наиболее вероятные гены-кандидаты – те, которые окажутся в обоих списках.
Степень восприимчивости к определенным лекарственным препаратам и эффективность их применения варьирует в широких пределах. При одном и том же заболевании подходящий для конкретного индивидуума препарат часто подбирают методом проб и ошибок. Кроме потери времени такой подход иногда наносит непоправимый вред здоровью. В настоящее время для большого количества лекарственных средств разработаны системы маркеров на основе SNP, позволяющие a priori (до опыта) предсказать реакцию индивидуального организма на то или иное химическое вещество. Ассоциации отдельных аллельных вариантов ДНК-маркеров с особенностями биохимических реакций являются основой индивидуальной терапии (Рисунок IX, 10).

Рисунок IX, 8. В микросателлитных локусах единицей изменчивости является группа нуклеотидов.

Рисунок IX, 9. Вмононуклеотидных полиморфных сайтах (SNP) единицей изменчивости является один нуклеотид.
Таблица IX, 1. Сравнение основных характеристик SNP и микросателлитов.

Рисунок IX, 10. Принцип подбора индивидуальной терапии на основе полиморфизма мононуклеотидных повторов - SNP.
Контрольные вопросы и задания к главе IX
1. К какой группе наследственных заболеваний можно отнести муковисцедоз?
2. Может ли у гетерозиготы по мутации гена SPTA1 быть наследственный сфероцитоз?
3. Какое наследственное заболевание вызвано накоплением гепарансульфата?
4. Почему число возможных аллелей SNP четыре?
Дополнительная литература к главе IX
Н.П. Бочков. Клиническая генетика // М.: Гэотар-Мед. 2002. – 457 С.






