بيلة الفينيل كيتون هي مرض وراثي شديد إلى حد ما، وتتركز الشدة الرئيسية لمظاهره في المقام الأول على الجهاز العصبي. تحدث بيلة الفينيل كيتون، التي توجد أعراضها في أغلب الأحيان بين الفتيات، نتيجة لانتهاك استقلاب الأحماض الأمينية، وبالنظر إلى الأضرار التي لحقت بالجهاز العصبي المركزي، تنخفض مظاهره إلى ضعف النمو العقلي.
وصف عام
في شكله الكلاسيكي، الذي ينطبق على معظم الحالات، ترتبط بيلة الفينيل كيتون، والتي تُعرف أيضًا بشكل شائع باسم قلة الفينيلبيروفيك، بانخفاض حاد في نشاط إنزيم الكبد. على وجه الخصوص، هو فينيلانين-4-هيدروكسيلاز. تتطلب الحالة الطبيعية للجسم تحفيز تحويل الفينيل ألانين إلى تيروزين. تتميز 1% من الحالات بحدوث شكل غير نمطي من بيلة الفينيل كيتون، والذي يحدث بسبب طفرات في نوع آخر من الجينات المسؤولة عن عملية ترميز الإنزيمات. وراثة المرض يحدث وفقا لنمط جسمي متنحي.
أما العمليات التي تحدث أثناء المرض المعني فهي على النحو التالي. يؤدي حدوث كتلة استقلابية مميزة إلى تنشيط المسارات الجانبية لعملية استقلاب الفينيل ألانين، مما يؤدي إلى تراكم المشتقات السامة من تأثيره. وتشمل هذه المشتقات على وجه الخصوص أحماض فينيللاكتيك وفينيلبيروفيك، والتي لا تتشكل عمليا في الحالة الطبيعية للجسم. كما أنها تؤثر على الجهاز العصبي المركزي، مما يسبب اضطرابات في استقلاب البروتين، استقلاب البروتين الدهني واستقلاب البروتين السكري. في الوقت نفسه، تنشأ اضطرابات في نقل الأحماض الأمينية، واضطرابات في استقلاب السيروتونين والكاتيكولامينات، كما تصبح عوامل الفترة المحيطة بالولادة ذات صلة.
بالإضافة إلى ذلك، يبدأ أيضًا في التشكل خلات أورثو فينيل وفينيل إيثيل أمين، والتي تكون غائبة عمليًا بشكل طبيعي. فائضها يثير اضطرابات في استقلاب الدهون التي تحدث في الدماغ. ومن المفترض أن هذا هو ما يسبب حالة التراجع التدريجي في الذكاء لدى المرضى المصابين بهذا المرض، والتي قد تصل حتى إلى البلاهة. وبشكل عام، فإن الآلية النهائية التي يحدث بموجبها انتهاك في تطور وظائف المخ في حالة مرض بيلة الفينيل كيتون، وهو مرض يتعلق بالجسم، ليست واضحة.
بيلة الفينيل كيتون: أعراض المرض
هنا، أولا وقبل كل شيء، من المهم أن نلاحظ أن الأسابيع الأولى من حياة الطفل لا تسمح بتحديد خارجي لهذا المرض. تظهر علاماته الأولى بعد شهرين إلى ستة أشهر من ولادة الطفل. ويصبح خاملاً ويفتقر إلى الاهتمام بالظروف المحيطة به وبالعالم بشكل عام. يصبح الطفل أيضًا مضطربًا وتتأثر قوة العضلات. تظهر القيء والتشنجات ومشاكل الجلد الشديدة. تشير الأكزيما على وجه الخصوص إلى شكل حاد أو مزمن من مرض التهابي وغير معدي يتشكل فيه الطفح الجلدي. طبيعة المرض حساسية. تشمل المظاهر الإضافية للأعراض إحساسًا بالحرقان وحكة شديدة في الجلد. يرتبط الميل إلى الانتكاس أيضًا، أي بعودة ظهور الأعراض بعد فترة هدوء مؤقتة نسبية.
الشهر السادس يسمح لك بتحديد تأخر نمو الطفل. في الوقت نفسه، يتم فقدان القدرة على تركيز النظرة على العناصر الفردية، ويتوقف الطفل عن التعرف على والديه. لا يوجد أي رد فعل تجاه الألعاب الملونة/المشرقة. من المهم أن تبدأ العلاج على الفور، وإلا فإن التخلف النمائي سوف يخضع تدريجيا فقط لتقدم في العمليات ذات الصلة به.
يتميز النمو الجسدي للأطفال المرضى على المستوى الجسدي باضطرابات أقل منه على المستوى العقلي. قد يكون محيط الرأس أصغر قليلاً من المعتاد. تظهر الأسنان لاحقًا، وبعد ذلك يبدأ الطفل بالجلوس والمشي. يتضمن اعتماد وضعية الوقوف عند هؤلاء الأطفال نشر الساقين على نطاق واسع، بالإضافة إلى ثنيهما عند الركبتين ومفاصل الورك، بينما يتم خفض الكتفين والرأس. أما خصائص المشي عند الأطفال المرضى فهي تتميز بالتمايل والخطوات الصغيرة. يجلس الأطفال وأرجلهم مدسوسة تحتهم، وذلك بسبب قوة العضلات الكبيرة التي يعانون منها.
كما يختلف الأطفال في مظهرهم المميز بشعر أشقر، وبشرتهم بيضاء بالكامل، بدون تصبغ، وعيونهم فاتحة. نظرًا للبياض المفرط للجلد، غالبًا ما يكون مغطى بطفح جلدي عند الأطفال، وهو ما يفسره حساسيته الخاصة لتأثيرات الأشعة فوق البنفسجية.
من بين المظاهر الرئيسية المميزة لبيلة الفينيل كيتون، يمكن أيضًا تسليط الضوء على رائحة "الفأر" المميزة، والتي تكون ممكنة في بعض الحالات، والتي تختفي مع تقدم العمر. المظاهر الواضحة هي زرقة الأطراف، ورسم الجلد (تغير في لون الجلد المحلي الذي يحدث تحت التأثير الميكانيكي عليه)، والتعرق. في كثير من الأحيان، بين المرضى الذين يعانون من بيلة الفينيل كيتون، بالإضافة إلى الأعراض المذكورة، وجود الإمساك المتكرر، والهزات (أي يرتجف)، وفقدان التوازن والاضطراب في شكل ضعف التنسيق بين الحركات.
تشخيص بيلة الفينيل كيتون
من المهم، كما أشرنا بالفعل، تشخيص المرض مبكرًا، مما سيتجنب تطوره ويؤدي إلى عدد من العواقب الوخيمة التي لا رجعة فيها. لهذا السبب، في مستشفيات الولادة، بعد 4-5 أيام من الحياة (للمواليد الجدد) يتم أخذ الدم للتحليل. بالنسبة للأطفال المبتسرين، يتم سحب الدم لفحص بيلة الفينيل كيتون (PKU) في اليوم السابع.
يتضمن الإجراء أخذ الدم الشعري بعد ساعة من الرضاعة، ويتم تشريب شكل خاص به. ويتطلب التركيز الذي يزيد عن 2.2% فينيل ألانين في دم الطفل إرساله هو ووالديه للفحص إلى أحد مراكز الجينات الطبية. هناك، يتم إجراء مزيد من الفحص، وفي الواقع، توضيح التشخيص.
أسباب بيلة الفينيل كيتون
يمكن أن تحدث بيلة الفينيل كيتون بسبب العوامل التالية:
- زواج الأقارب، الذي، بالإضافة إلى الأمراض الأخرى، يزيد من احتمال إنجاب طفل مصاب بهذا المرض؛
- طفرة جينية (أي تغيرها) حدثت لسبب أو لآخر في منطقة توطين الكروموسوم 12.

علاج بيلة الفينيل كيتون
الطريقة الوحيدة للعلاج هي تنظيم العلاج الغذائي في الوقت المناسب، وهو أمر مطلوب من الأيام الأولى من الحياة. وهو يتألف من محدودية حادة في مادة الفينيل ألانين الموجودة في بعض الأطعمة. وبالتالي، يتم استبعاد جميع منتجات البروتين. بالنظر إلى المدة والاستبعاد الكامل للفينيل ألانين من الطعام، يصبح من الممكن انهيار البروتينات الخاصة بالفرد، مما يؤدي إلى استنفاد جسم المريض. لهذا السبب، يتم تعويض الحاجة إلى البروتين عن طريق مخاليط الأحماض الأمينية وهيدرات البروتين.
ومع انخفاض تركيز الفينيل ألانين في الدم إلى مستوياته الطبيعية، تتم إضافة المنتجات الحيوانية تدريجياً إلى النظام الغذائي. يتم إضافة الفواكه والخضروات الموسمية المختلفة والدهون النباتية والحيوانية وكذلك الكربوهيدرات إلى النظام الغذائي. وبطبيعة الحال، ينبغي الاستمرار في الحفاظ على السيطرة على محتوى الفينيل ألانين. ويجب تزويد الجسم بهذا الحمض الأميني بالكمية المناسبة للتطور والنمو الطبيعي للطفل، باستثناء إمكانية تراكمه في الأنسجة.
يتم اتباع النظام الغذائي الصارم لمدة خمس سنوات على الأقل. أما بالنسبة لكبار السن، فإن قابلية الجهاز العصبي لتأثيرات الفينيل ألانين، وكذلك تأثيرات منتجات تحلله، تقل بشكل كبير. إذا تم اتباع التدابير المطلوبة، بحلول سن 12-14 سنة، سيكون الطفل قادرا على التحول بحرية إلى نظام غذائي عادي.
يشار إلى أن العلاج الدوائي لهذا المرض له طبيعة متلازمية. ويشمل استخدام الأدوية التي تهدف إلى القضاء على النوبات، بما في ذلك الأدوية التي لها تأثير محفز للنشاط الفكري. يُطلب من الأطفال الخضوع لدورة من العلاج الطبيعي والتدليك. بالإضافة إلى ذلك، يتم تعيين الدروس التي تعزز تطوير المنطق.
يتم تشخيص بيلة الفينيل كيتون من قبل طبيب الأطفال وعلم الوراثة على أساس اختبارات خاصة بالاشتراك مع الأعراض المميزة العامة.
هل كل ما ورد في المقال صحيح من الناحية الطبية؟
أجب فقط إذا كان لديك معرفة طبية مثبتة
الأمراض ذات الأعراض المشابهة:
الوذمة الدماغية هي حالة خطيرة تتميز بالتراكم المفرط للإفرازات في أنسجة العضو. ونتيجة لذلك، يزداد حجمه تدريجياً ويزداد الضغط داخل الجمجمة. وكل هذا يؤدي إلى اضطراب الدورة الدموية في العضو وموت خلاياه.
الإرهاق هو حالة لا يواجهها البالغون فحسب، بل يواجهها الأطفال أيضًا اليوم. ويتميز بانخفاض النشاط والنعاس وضعف الانتباه والتهيج. علاوة على ذلك، يعتقد الكثير من الناس أن الإرهاق في العمل ليس مشكلة خطيرة، وأنه يكفي الحصول على نوم جيد ليلاً حتى تختفي هذه المشكلة. في الواقع، من المستحيل التخلص من مثل هذا الاضطراب عن طريق النوم لفترة طويلة. والعكس صحيح - فالرغبة المستمرة في النوم وعدم القدرة على استعادة القوة بعد النوم هي الأعراض الرئيسية للإرهاق.
الورم الغدي جارات الدرق هو تكوين حميد صغير يتراوح حجمه من 1 إلى 5 سم، ويمكنه تصنيع هرمون الغدة الدرقية بشكل مستقل، مما يسبب أعراض فرط كالسيوم الدم لدى الشخص. تقع الغدد جارات الدرق على السطح الخلفي للغدة الدرقية، والغرض الرئيسي منها هو إنتاج هرمون الغدة الدرقية، الذي يشارك في استقلاب الكالسيوم والفوسفور في الجسم. يؤدي الورم الحميد إلى إنتاج هرمون الغدة الدرقية أكثر من اللازم، مما يسبب أعراض هذا المرض.
استسقاء الرأس، والذي يُعرف أيضًا باسم الاستسقاء الدماغي، هو مرض يزداد فيه حجم البطينات في الدماغ، وغالبًا ما يصل إلى حجم مثير للإعجاب. استسقاء الرأس، الذي تظهر أعراضه بسبب الإفراط في إنتاج السائل النخاعي (السائل النخاعي بين البطينين المتصلين بالدماغ) وتراكمه في منطقة تجاويف الدماغ، يحدث بشكل رئيسي عند الأطفال حديثي الولادة، ولكن هذا المرض يحدث أيضًا في حالات الاعتلال في الفئات العمرية الأخرى.
استسقاء الرأس (syn. الاستسقاء) في الدماغ عند الأطفال هو مرض يتميز بحقيقة أن كمية زائدة من السائل النخاعي، المعروف أيضًا باسم السائل النخاعي، تتجمع في تجاويفها الداخلية وتحت السحايا. هناك أسباب كثيرة لتكوين المرض، وقد تختلف تبعا للعمر الذي تشكلت فيه الأمراض. العوامل الاستفزازية الأكثر شيوعًا هي العمليات المعدية والأورام والتشوهات الخلقية وإصابات الولادة.
بيلة الفينيل كيتون (PKU) هو مرض وراثي نادر يرتبط باضطراب في استقلاب الفينيل ألانين (حمض أميني بروتيني المنشأ) الذي يدخل الجسم كجزء من الأطعمة البروتينية. يسبب تراكم مادة الفينيل ألانين ومشتقاته السامة في الجسم، مما يؤدي إلى تلف الجهاز العصبي المركزي.
| التصنيف الدولي للأمراض-10 | E70.0 |
|---|---|
| التصنيف الدولي للأمراض-9 | 270.1 |
| الأمراضDB | 9987 |
| ميدلاين بلس | 001166 |
| الطب الإلكتروني | بيد/1787 ديرم/712 |
| مش | D010661 |
| أوميم | 261600 261630 |
معلومات عامة
كانت بيلة الفينيل كيتون معروفة منذ عام 1934، ولكن بسبب الافتقار إلى تشخيصات عالية الجودة، تم اكتشاف المرض فقط في المرحلة التي تصبح فيها اضطرابات النمو العقلي غير قابلة للشفاء نتيجة للتأثيرات السامة على الدماغ. ولهذا السبب حصل المرض على اسم ثانٍ - قلة فينيلبيروفيك.
وفقا للإحصاءات، يتم ملاحظة اضطرابات التمثيل الغذائي للفينيل ألانين في كثير من الأحيان عند الفتيات. إنه أكثر شيوعا في تركيا ودول أوروبا الشمالية (باستثناء فنلندا)، وهو نادر للغاية بين ممثلي العرق الزنجي. بما أن بيلة الفينيل كيتون هي مرض يتم فيه توريث الجين المعيب من كلا الوالدين، فإن زواج الأقارب يزيد من احتمالية إنجاب أطفال يعانون من هذا المرض.
تم إجراء أول علاج ناجح في مستشفى الأطفال في برمنغهام (إنجلترا) في الخمسينيات. القرن العشرين، لكن طريقة العلاج هذه أصبحت فعالة حقًا بعد تقديمها في أوائل الستينيات. التشخيص المبكر.
صِنف
ويتحول الفينيل ألانين، تحت تأثير الإنزيمات الموجودة في الجسم، إلى التيروزين، وهو حمض أميني يفرز من الجسم. اعتمادًا على الخلل الجيني الذي يمنع إنزيمًا معينًا، يتم تمييز ما يلي:
- بيلة الفينيل كيتون من النوع الأول (الكلاسيكية أو الشديدة). يحدث بسبب طفرة جينية تعطل إنتاج إنزيم الكبد فينيل ألانين هيدروكسيلاز وتحويل الفينيل ألانين إلى تيروزين.
- بيلة الفينيل كيتون من النوع الثاني (غير نمطية). يتميز بوجود خلل جيني يسبب نقص إنزيم ثنائي هيدروبيوبترين المختزل. وبسبب هذا العامل، يتم تعطيل استعادة نشاط المركب العضوي اللازم لتحويل الفينيل ألانين. هناك أيضًا انخفاض في محتوى فيتامين ب9 في السائل النخاعي ومصل الدم، وهو أمر ضروري للاستفادة من الأحماض الأمينية.
- بيلة الفينيل كيتون من النوع الثالث (غير نمطية). وينجم عن نقص المحفز اللازم لتخليق رباعي هيدروبيوبترين (اللازم لتحويل الفينيل ألانين إلى تيروزين).
- Primapterinuria هو شكل غير نمطي يتم ملاحظته في الأشكال الخفيفة من فرط فينيل ألانين الدم. إن الخلل الإنزيمي لهذا النوع من بيلة الفينيل كيتون غير واضح حاليًا، لكن هذا الشكل من المرض يتميز بوجود كمية كبيرة من البريمابترين ومشتقاته في البول، كما أن كمية مستقلبات الناقلات العصبية في السائل النخاعي لا تنحرف عن القاعدة.
يتم أيضًا تحديد بيلة الفينيل كيتون الأمومية، والتي يتم ملاحظتها في ذرية النساء المصابات ببيلة الفينيل كيتون (PKU) والذين لا يتبعون نظامًا غذائيًا متخصصًا. لم تتم دراسة التسبب في هذا النوع من المرض بشكل كامل، ولكن لوحظ أنه بدون المراقبة المستمرة لمستويات الفينيل ألانين لدى الأطفال حديثي الولادة، يتم اكتشاف عدد من التغيرات المرضية:
- عدم كفاية وزن الدماغ.
- تضخم البطينات في الدماغ (تضخم البطين) ؛
- نقص تنسج (التخلف) في المادة البيضاء وتأخر تكون الميالين.
يسبب هذا النوع من بيلة الفينيل كيتون تسممًا مزمنًا للجنين ويؤدي إلى تخلف عقلي لدى الطفل.
أسباب التطوير
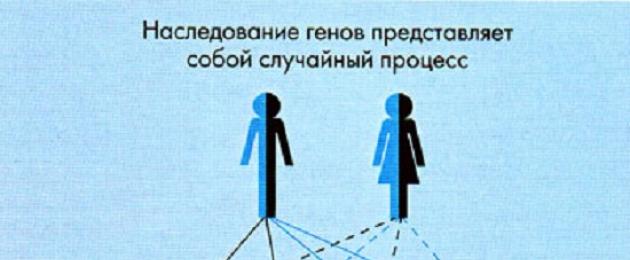
يحدث قلة الفينيلبيروفيك عندما يكون الجين المرضي موجودًا في الأم والأب. نوع الميراث هو جسمي متنحي، أي أنه لا يعتمد على جنس الطفل. تبلغ احتمالية الإصابة بالمرض عند الأطفال حديثي الولادة الذين يكون آباؤهم حاملين للجين المعيب 25٪.
المرضية
نتيجة لاضطراب في استقلاب الفينيل ألانين الناجم عن خلل في الجينات وتحوله لاحقًا إلى تيروزين، تتراكم في الجسم مشتقات سامة من هذا الحمض الأميني (أحماض فينيلبيروفيك وفينيللاكتيك وفينيل أسيتيك)، والتي توجد عادة بكميات ضئيلة. . كما يتم تكوين أورثو فينيل أسيتات وفينيل إيثيل أمين، والتي لا توجد عادة، مما يعطل استقلاب الدهون في الدماغ، مما يسبب انخفاضًا تدريجيًا في الذكاء.
العوامل التي تؤثر على تطور علم الأمراض هي:
- اضطرابات استقلاب الأحماض الأمينية.
- اضطرابات الميالين.
- انتهاك تخليق البروتينات الدهنية ،
- انخفاض مستوى الناقلات العصبية (الأدرينالين والسيروتونين وما إلى ذلك).
أعراض
بعد الولادة مباشرة، لا يعاني الأطفال المصابون ببيلة الفينيل كيتون I من أعراض المرض، على الرغم من أن لديهم عادةً عددًا محددًا من العلامات الخارجية:
- بشرة جافة وبيضاء (تفتقر تمامًا إلى التصبغ).
- عيون زرقاء.
- شعر ذو ظلال خفيفة.
في عمر 2-6 أشهر تظهر العلامات الأولى للمرض:
- الخمول (لا يحاول التدحرج أو الجلوس)؛
- التصور السلبي للبيئة (لا يتفاعل مع الأم، لا يستجيب بابتسامة)؛
- زيادة التهيج.
- تأخر التطور النفسي الحركي.
القيء المتكرر الشديد والقلق، والنوبات ممكنة.
إذا لم يتم اكتشاف المرض في الوقت المناسب وإدخال الأطعمة البروتينية في نظام الطفل الغذائي، تبدأ الأعراض في الزيادة. في مثل هؤلاء الأطفال، تظهر الأسنان في وقت متأخر، ويقل حجم الجمجمة إلى حد ما. يبدأون في الجلوس والمشي في وقت متأخر عن أقرانهم، وتعبيرات وجوههم غير معبرة، وفي عام واحد لا يتمكنون من التعبير عن مشاعرهم بصوتهم، ولا يفهمون كلام البالغين. احتمالية تأخر النمو.
وبما أن الفينيل ألانين لا يتحول في الجسم، فإنه يفرز في العرق والبول، ولهذا السبب يكون لدى المرضى رائحة عفنة أو "موسية".
تتجلى بيلة الفينيل كيتون عند الأطفال أيضًا في أوضاع ومشية غريبة، نظرًا لزيادة قوة العضلات لدى هؤلاء المرضى. في وضعية الوقوف، تكون أرجل الطفل متباعدة بشكل واسع ومثنية عند الركبتين ومفاصل الورك، ويتم خفض الرأس والكتفين. المشية متأرجحة والخطوات صغيرة. يجلسون في وضع الخياط (أرجلهم مطويّة ومتقاطعة).
يتجلى قلة الفينيلبيروفيك بعد سن الثالثة في:
- زيادة الإثارة والتعب.
- الاضطرابات السلوكية
- اضطرابات ذهانية
- التخلف العقلي.
غالبًا ما يتم ملاحظة الأكزيما وتصلب الجلد والتهاب الجلد.
وبدون علاج، تتفاقم حالة المرضى. التشخيص والعلاج في الوقت المناسب يمكن أن يمنع اضطرابات نمو الطفل.
التشخيص
يمكن تشخيص المرض:
- اختبار شبه كمي يسمح لك بتحديد الكمية التقريبية للفينيل ألانين في الدم؛
- التحديد الكمي باستخدام الكواشف التي تكشف كمية الفينيل ألانين في بقع الدم المجففة.
لإجراء هذا الفحص، يتم أخذ الدم من كعب جميع الأطفال بعد الرضاعة في مستشفى الولادة في يوم الخروج أو في اليوم الخامس من الحياة.

يشمل التشخيص أيضًا اختبار البول (اختبار القطع)، ولكنه يكون مفيدًا فقط بعد 10-12 يومًا من حياة الطفل. يظهر حمض فينيلبيروفيك بلون أزرق مخضر في البول عند إضافة كلوريد الحديديك إلى البول.
ومن الممكن أيضًا تحديد نشاط إنزيم فينيل ألانين هيدروكسيلاز في مادة الخزعة واكتشاف الطفرات في الجين.
طرق مثل:
- اختبار جوثري، يعتمد على إضافة الدم إلى مزرعة بكتيرية تنمو بسرعة في وسط الفينيل ألانين؛
- كروماتوغرافيا الطبقة الرقيقة للأحماض الأمينية الموجودة في مصل الدم؛
- قياس الفلور، مما يجعل من الممكن الكشف عن جرعات صغيرة من الفينيل ألانين بسبب الأشعة فوق البنفسجية.
يتم تشخيص النوعين 2 و3 من بيلة الفينيل كيتون باستخدام بيوبترينات البول، واختبار الإجهاد رباعي هيدروبيوبترين عن طريق الفم، والدراسات الأنزيمية.
يمكن أيضًا اكتشاف بيلة الفينيل كيتون باستخدام التحليل الجيني، والذي يتم إجراؤه عادةً إذا كان هناك أفراد في العائلة مصابون بهذا التشخيص.
علاج
إذا تم تشخيص بيلة الفينيل كيتون في الوقت المناسب، فإن علاج بيلة الفينيل كيتون يتكون من اتباع نظام غذائي متخصص يحد من تناول الأطعمة التي تحتوي على الفينيل ألانين. وبما أن جميع مصادر البروتين الطبيعية تحتوي على حوالي 4% من الفينيل ألانين، فقد تم استبدالها بمنتجات صناعية تحتوي على أحماض أمينية أخرى. يتم وصف النظام الغذائي الأكثر فعالية قبل الأسبوع الثامن من العمر.
يوصف للرضع تركيبات خالية تمامًا من اللاكتوز تعتمد على هيدروليزات بروتين الحليب. حليب الثدي مقبول بكميات محدودة.
على الرغم من أن الأطباء أوصوا سابقًا باتباع نظام غذائي فقط حتى نهاية نمو الدماغ (سن 20 عامًا)، إلا أن ارتفاع مستويات الفينيل ألانين بعد التوقف عن النظام الغذائي يسبب مشاكل نفسية لدى العديد من الأشخاص. تتجلى بيلة الفينيل كيتون لدى البالغين في قلة الحافز والأرق وعدم التركيز والاندفاع وما إلى ذلك، لذلك يوصى باتباع النظام الغذائي مدى الحياة.
القيود الغذائية التي يتم إدخالها بعد عامين لا يمكن إلا أن تقلل من شدة الأعراض.
تستخدم الأدوية التالية أيضًا للعلاج:
- ينتمون إلى مجموعة منشط الذهن (نوتروبيل، وما إلى ذلك). أنها تحفز النشاط العقلي وتنشط الوظائف المعرفية.
- تحتوي على الفيتامينات والأحماض الأمينية والبروتينات ("أفينيلاك"، وما إلى ذلك).
الأشكال غير النمطية من بيلة الفينيل كيتون غير قابلة للعلاج بالنظام الغذائي وتتطلب علاجًا دوائيًا إضافيًا، والذي يتضمن:
- ثنائي هيدروبيوبترين، لتعويض نقص حمض الفوليك؛
- الليفودول له تأثير على الجهاز العصبي المركزي.
يتم وصف مادوبار أو ناكوم للمرضى الذين يحتاجون إلى تصحيح إضافي للناقلات العصبية.
توصف للبالغين مستحضرات عشبية تحتوي على هيدروكسيلاز الفينيل ألانين.
وقاية
نظرًا لأن بيلة الفينيل كيتون مرض وراثي، فلا يمكن منع تطوره بشكل كامل. تهدف التدابير الوقائية إلى منع الاضطرابات الشديدة التي لا رجعة فيها في نمو الدماغ من خلال التشخيص في الوقت المناسب والعلاج الغذائي.
يُنصح العائلات التي سبق أن ظهرت فيها حالات من هذا المرض بإجراء تحليل جيني يمكنه التنبؤ بالتطور المحتمل لبيلة الفينيل كيتون لدى الطفل.
مرض فهلنغ أو بيلة الفينيل كيتون هو مرض وراثي لاستقلاب الأحماض الأمينية. وهذا هو فشل عمليات استيعاب فينيل ألانين، التي تسبب منتجاتها السامة "تسمم" الجهاز العصبي والدماغ، مما يؤدي إلى تطور اضطرابات وتشوهات خطيرة. ومن خلال التشخيص المبكر والالتزام الصارم بنظام غذائي خاص، يمكن تجنب العواقب الوخيمة لتطور هذا المرض النادر.
كيف تتطور بيلة الفينيل كيتون؟
يسمى المرض الوراثي الخلقي الناجم عن اضطراب استقلاب الأحماض الأمينية ببيلة الفينيل كيتون. الأسماء الأخرى لهذا المرض هي مرض فيلينغ (على اسم الطبيب الذي وصف المرض لأول مرة) أو قلة فينيلبيروفيك (العواقب الرئيسية لتطور المرض هي الأضرار الجسيمة للجهاز العصبي المركزي والدماغ، مما يسبب تأخر النمو والتخلف العقلي).
في روسيا، يتم تسجيل بيلة الفينيل كيتون بتكرار حالة واحدة لكل 5-10 آلاف مولود جديد، في تركيا هذه النسبة هي 1: 2500، في اليابان وبعض الدول الأوروبية - 1: 100000، في البلدان الأفريقية لا يحدث المرض عمليا. وفقا للإحصاءات، ترث الفتيات علم الأمراض مرتين أكثر من الأولاد.في فترة حديثي الولادة، لا توجد مظاهر سريرية للمرض، ولكن بسبب تناول الفينيل ألانين في الجسم مع الطعام، تحدث مظاهر بيلة الفينيل كيتون في الأشهر الستة الأولى من الحياة.
في مرض فيلينغ، يؤدي نقص إنزيم الكبد فينيل ألانين 4-هيدروكسيلاز إلى حدوث اضطرابات في استقلاب الفينيل ألانين الذي يتم توفيره مع الأطعمة البروتينية. تحدث العمليات التالية:
- يتم تشغيل مسارات جانبية لأكسدة الفينيل ألانين، حيث تتراكم مشتقاته السامة في الجسم - أحماض فينيل أسيتيك وفينيلكتيك وفينيل بيروفيك.
- هناك نقص في التيروزين (حمض أميني، نتاج التمثيل الغذائي الطبيعي للفينيل ألانين)، الذي يشارك في وظائف الغدة النخامية والغدد الكظرية والغدة الدرقية، وإنتاج الميلانين، الذي ينظم الشهية وعمليات الدهون الودائع.
- تتطور الاضطرابات في استقلاب البروتينات الدهنية والبروتينات السكرية والبروتينات.
- هناك اضطراب في نقل الأحماض الأمينية.
- ينتهك تبادل الكاتيكولامينات والسيروتونين.
- يتم تشكيل فائض من فينيثيلامين وأورثوفينيل أسيتات، مما يثير اضطرابات استقلاب الدهون في الدماغ.
- تدمير آلية نقل النبضات بين خلايا الجهاز العصبي بسبب النقص الناتج في الناقلات العصبية.
الأسباب
تتطور متلازمة بيلة الفينيل كيتون عند الأطفال في 98٪ من الحالات نتيجة لطفرة تقع على الذراع الطويلة للكروموسوم 12 من الجين الذي يشفر كمية هيدروكسيلاز فينيل ألانين (مما يؤدي إلى نقص هذا الإنزيم). علم الأمراض الوراثي ذو طبيعة وراثية متنحية للميراث، أي أنه من أجل تطور المرض، يجب أن يرث الوليد نسخة من الجين المتحور المعيب من كل من الأب والأم، اللذين هما حامله المتغاير الزيجوت. ويرجع ذلك إلى انخفاض معدل انتشار علم الأمراض.
الأشكال والأنواع
وتمثل بيلة الفينيل كيتون الكلاسيكية من النوع 1، والتي تتطور نتيجة وراثة جين متحور من كلا الوالدين، 98-99٪ من جميع الحالات المسجلة. الأشكال غير النمطية الأخرى من المرض التي لا يمكن علاجها بالعلاج الغذائي، ولكنها تحدث بمظاهر سريرية مماثلة، هي:
- بيلة الفينيل كيتون من النوع 2، والتي تتطور نتيجة لنقص إنزيم ديهيدروبترين ريدكتيز.
- بيلة الفينيل كيتون من النوع 3، الناتجة عن نقص رباعي هيدروبيوبترين.
أعراض بيلة الفينيل كيتون
في فترة حديثي الولادة (الأسابيع القليلة الأولى بعد الولادة)، لا توجد أعراض بيلة الفينيل كيتون. تظهر الأعراض الأولى غير النوعية بعد شهرين إلى ستة أشهر من بدء الطفل في تغذية حليب الثدي أو بدائله، وذلك لأن البروتين يزود الجسم بانتظام بالطعام. مع نمو الطفل، يتم ملاحظة العلامات السريرية التالية للمرض:
- العمر 6 أشهر: الضعف والخمول وقلة الانفعالات والاهتمام بما يحدث حولها. - عدم استجابة الطفل لوالديه وكلامهم، للألعاب ذات الألوان الزاهية، ويتميز بعدم القدرة على تركيز نظره وعدم تعبير تعبيرات الوجه. في بعض الحالات - القلق، فرط الاستثارة، واضطرابات توتر العضلات (نقص التوتر العضلي أو خلل التوتر العضلي). القيء، المتلازمة المتشنجة، الأكزيما الجلدية.
- العمر من 6 أشهر إلى سنة: تأخر في النمو الحركي النفسي - انخفاض النشاط، قلة محاولات تعلم الجلوس والوقوف. يؤدي وجود منتجات تحلل الفينيل ألانين السامة في البول والعرق إلى ظهور رائحة عفن معينة من الجسم. العلامات الجسدية هي انحرافات طفيفة عن المعتاد في حجم الرأس؛ فعند الوقوف يضع الطفل ساقيه على نطاق أوسع من المسافة المطلوبة، وعند المشي يتمايل، وعند الجلوس يميل إلى وضع ساقيه تحته.
- العمر أكثر من 12 شهرًا: تأخر التسنين، صغر الرأس، نقص تنسج المينا، تأخر تطور الكلام. في بعض الحالات، تكون نوبات الصرع ممكنة. مع التقدم في السن، يتطور انخفاض ضغط الدم، والتهاب الجلد، والميل إلى الإمساك. رعاش الأطراف، واضطرابات التنسيق، ومشية التقطيع. زرقة محتملة في الأطراف ، تصوير الجلد. بسبب نقص التيروزين، يكون لدى الأطفال المرضى مظهر محدد - شعر أشقر، أبيض، بدون تصبغ، بشرة حساسة للأشعة فوق البنفسجية، عيون فاتحة.
- العمر 3-4 سنوات: تخلف عقلي عميق (البلاهة)، قلة النطق.
تشخيص بيلة الفينيل كيتون
يُطلق على إجراء فحص المولود الجديد للكشف عن بيلة الفينيل كيتون أو غيرها من الأمراض الوراثية الشديدة اسم فحص حديثي الولادة وهو إلزامي في الاتحاد الروسي. وينظم هذا الموضوع أمر وزارة الصحة رقم 316 المؤرخ 30 كانون الأول (ديسمبر) 1993. يتم تنفيذ الإجراء في مستشفيات الولادة. في الأطفال الناضجين، يتم أخذ الدم من الكعب للتحليل في اليوم الرابع من الحياة، عند الأطفال المبتسرين - في اليوم السابع. يتم تطبيق المادة على نموذج اختبار يتم إرساله إلى المختبر حيث يتم إجراء الاختبارات الجينية.
في حالة وجود فرط فينيل ألانين الدم (أكثر من 900 ميكرومول/لتر)، يتم إرسال الطفل لإجراء الاختبارات الجينية. للتأكد من تشخيص بيلة الفينيل كيتون عند الأطفال حديثي الولادة، يتم قياس تركيز التيروزين والفينيل ألانين في الدم، وفحص نشاط هيدروكسيلاز الفينيل ألانين (إنزيم الكبد)، ويتم إجراء اختبار البول البيوكيميائي لتحديد مستوى مستقلبات الكاتيكولامين والأحماض الكيتونية. . من الممكن إجراء تخطيط كهربية الدماغ (EEG) والتصوير بالرنين المغناطيسي (التصوير بالرنين المغناطيسي) للدماغ، والفحص من قبل طبيب أعصاب، والبرنامج المشترك وتشخيص الحمض النووي.
علاج
يتم علاج مرض بيلة الفينيل كيتون في روسيا من خلال العلاج الغذائي، مما يحد من استهلاك الأطعمة البروتينية. يقوم العالم بتطوير أدوية تسمح لك بالتحكم في مستويات الفينيل ألانين دون اتباع نظام غذائي صارم. تعتبر مجالات البحث الواعدة:
- استخدام إنزيم النبات فينيل ألانين لياز؛
- العلاج الجيني الذي يهدف إلى تصحيح الجين المتحور الذي يسبب اضطرابات في استقلاب الأحماض الأمينية؛
- محاولات لإدخال جين فينيل ألانين هيدروكسيلاز إلى خلايا الكبد المصابة.
المراقبة المستمرة من قبل الأطباء (طبيب أطفال، طبيب أعصاب)، والالتزام بنظام غذائي يحد من تناول البروتين منذ الولادة وحتى سن البلوغ هو الشرط الرئيسي للنمو الطبيعي للطفل الذي يعاني من الشكل الكلاسيكي لبيلة الفينيل كيتون. تتم مراقبة مستويات الفينيل ألانين بانتظام: في الأشهر الثلاثة الأولى من الحياة - مرة واحدة في الأسبوع، من 3 أشهر إلى سنة - مرة واحدة على الأقل في الشهر، من سنة إلى 3 سنوات - مرة كل شهرين، ثم - حسب توصية الطبيب (حوالي 4 مرات في السنة). يتم ضبط كمية البروتين التي يستهلكها المريض حسب العمر والحمل.

العلاج الغذائي
يعتمد النظام الغذائي لعلاج بيلة الفينيل كيتون على استبعاد البروتينات الحيوانية من النظام الغذائي للمريض. مع التقيد الصارم المستمر به، من الممكن منع تطور الاضطرابات الدماغية وضعف الكبد إذا تم تشخيص المرض في الأسابيع الأولى من الحياة. تشمل المنتجات المحظورة تمامًا ما يلي:
- جميع أنواع منتجات اللحوم والنقانق.
- جميع أنواع الأسماك والمأكولات البحرية.
- جبن؛
- بيض؛
- المكسرات.
- منتجات المخابز والحلويات؛
- منتجات الصويا؛
- الحبوب والرقائق.
يجب أن يحتوي النظام الغذائي اليومي للمريض على الخضار والفواكه والتوت والأعشاب. يُسمح باستهلاك العسل والسكر والنشا والأرز ودقيق الذرة والزبدة والزيت النباتي. يجب أن يكون عدد من المنتجات محدودًا جزئيًا. يمكن بعد إذن الطبيب المعالج استعمال كمية قليلة من:
- منتجات الألبان؛
- البطاطس والملفوف الأبيض.
- الخضار المعلبة.
تحتوي الأحماض الأمينية الضرورية لنمو ونمو الطفل على خلطات طبية خاصة منقاة من اللاكتوز تحتوي على الببتيدات (بروتينات الحليب التي تم تفكيكها بالفعل بواسطة الإنزيمات) والأحماض الأمينية الحرة (التريبتوفان والتيروزين والتورين والسيستين والهستيدين). يتم إنتاجها على شكل مساحيق مخففة بالماء أو حليب الثدي (بشرط أن تتبع الأم نظامًا غذائيًا خاصًا). هذه الخلطات والمنتجات الخاصة للأطفال من مختلف الأعمار هي:
- أفينيلاك.
- مدميل-FKU-0;
- التناظرية-SP؛
- أبونتي.
- بيرلافين.
- مينافين.
- لوفينيلاك.
- نوفيلان.
- تيترافين.
- سيمورجان.
العلاج الدوائي
حتى مع الاستلام المنتظم لهيدروليزات البروتين ومخاليط الأحماض الأمينية، يحتاج المرضى الذين يعانون من بيلة الفينيل كيتون إلى إدارة إضافية لمجمعات الفيتامينات (على سبيل المثال، المجموعة ب) والمركبات المعدنية (التي تحتوي على الحديد والفوسفور والعناصر النزرة الأخرى). يتم إجراء العلاج الدوائي للأعراض والمسببة للأمراض باستخدام.
34. بيلة الفينيل كيتون، الخلل البيوكيميائي، مظهر المرض، التشخيص، العلاج.
بيلة الفينيل كيتون في كبد الأشخاص الأصحاء، يتحول جزء صغير من الفينيل ألانين (∼10%) إلى فينيل لاكتات وفينيل أسيتيل جلوتامين. يصبح هذا المسار لتقويض الفينيل ألانين هو المسار الرئيسي عندما يتم تعطيل المسار الرئيسي - التحويل إلى التيروزين، المحفز بواسطة هيدروكسيلاز فينيل ألانين. يصاحب هذا الاضطراب فرط فينيل ألانين الدم وزيادة في محتوى الدم والبول من مستقلبات المسار البديل: فينيل بيروفات، فينيل أسيتات، فينيل لاكتات وفينيل أسيتيل جلوتامين.
يؤدي وجود خلل في هيدروكسيلاز الفينيل ألانين إلى مرض بيلة الفينيل كيتون (PKU). هناك نوعان من بيلة الفينيل كيتون:
بيلة الفينيل كيتون الكلاسيكية- مرض وراثي يرتبط بطفرات في جين الفينيل ألانين هيدروكسيلاز مما يؤدي إلى انخفاض نشاط الإنزيم أو تعطيله بالكامل.
في هذه الحالة، يزيد تركيز الفينيل ألانين في الدم بمقدار 20-30 مرة (عادة - 1.0-2.0 ملغم/ديسيلتر)، في البول - بنسبة 100-300 مرة مقارنة بالمعدل الطبيعي (30 ملغم/ديسيلتر). يصل تركيز الفنيل بيروفات والفينيللاكتات في البول إلى 300-600 ملجم/ديسيلتر مع غياب كامل للمستوى الطبيعي. تتمثل أشد مظاهر مرض بيلة الفينيل كيتون في ضعف النمو العقلي والجسدي، والمتلازمة المتشنجة، واضطرابات التصبغ. بدون علاج، لا يعيش المرضى أكثر من 30 عامًا. نسبة الإصابة بالمرض هي 1:10.000 مولود جديد.(فرط فينيل ألانين الدم المعتمد على الإنزيم المساعد) هو نتيجة لطفرات في الجينات التي تتحكم في استقلاب H4 BP.
المظاهر السريرية قريبة، ولكنها ليست متطابقة تمامًا، مع أعراض بيلة الفينيل كيتون الكلاسيكية. معدل الإصابة بالمرض هو 1-2 حالة لكل مليون مولود جديد. يعد H 4 BP ضروريًا لتفاعلات الهيدروكسيل ليس فقط فينيل ألانين، ولكن أيضًا التيروزين والتريبتوفان، لذلك، مع عدم وجود هذا الإنزيم المساعد، يتم انتهاك عملية التمثيل الغذائي لجميع الأحماض الأمينية الثلاثة، بما في ذلك تخليق الناقلات العصبية. يتميز المرض بضعف عصبي شديد والموت المبكر (PKU "الخبيث"). يمكن الوقاية من الضعف التدريجي للنمو العقلي والجسدي لدى الأطفال المصابين ببيلة الفينيل كيتون (PKU) عن طريق اتباع نظام غذائي منخفض جدًا في الفينيل ألانين أو يزيله. إذا بدأ هذا العلاج مباشرة بعد ولادة الطفل، فسيتم منع تلف الدماغ. يُعتقد أنه يمكن تخفيف القيود الغذائية بعد سن العاشرة (نهاية عملية تكون الميالين في الدماغ)، لكن في الوقت الحاضر يميل العديد من أطباء الأطفال نحو "نظام غذائي مدى الحياة". لتشخيص بيلة الفينيل كيتون (PKU).
استخدام الطرق النوعية والكمية للكشف عن المستقلبات المرضية في البول، وتحديد تركيز الفينيل ألانين في الدم والبول. يمكن اكتشاف الجين المعيب المسؤول عن بيلة الفينيل كيتون في حاملات الزيجوت غير المتجانسة ظاهريًا باستخدام اختبار تحمل الفينيل ألانين. وللقيام بذلك، يتم إعطاء الشخص ∼ 10 جرام من الفينيل ألانين على شكل محلول على معدة فارغة، ثم يتم أخذ عينات الدم على فترات كل ساعة، حيث يتم تحديد محتوى التيروزين. عادة، يكون تركيز التيروزين في الدم بعد تناول الفينيل ألانين أعلى بكثير منه في حاملات الجين المتغاير الزيجوت فيزيل كيتونوريا. يُستخدم هذا الاختبار في الاستشارة الوراثية لتحديد خطر إنجاب طفل مصاب. لقد تم تطوير مخطط فحص لتحديد الأطفال حديثي الولادة المصابين ببيلة الفينيل كيتون (PKU). تصل حساسية الاختبار إلى 100% تقريبًا. حاليًا، يمكن إجراء تشخيص الجين الطافر المسؤول عن PKU باستخدام طرق تشخيص الحمض النووي (تحليل التقييد وتفاعل البوليميراز المتسلسل).
بعد أن تعلمت ما هو نوع المرض - بيلة الفينيل كيتون، التي يتم تشخيصها خلال فترة حديثي الولادة، إذا تم اكتشافها، فمن الضروري البدء في العلاج على الفور. الكشف المبكر والعلاج يمكن أن يحقق نتائج إيجابية.
بيلة الفينيل كيتون، أو مرض فيلينغ، هو مرض خطير تم وصفه لأول مرة في عام 1934 من قبل العالم النرويجي فيلينغ. بعد ذلك قام فيلينج بفحص العديد من الأطفال وكشف عن وجود فينيل بيروفات في بولهم، وهو منتج تحلل للحمض الأميني فينيلانين الذي يتم تناوله مع الطعام، والذي لا يتحلل في جسم المرضى. بيلة الفينيل كيتون هي مرض يرتبط باضطراب التمثيل الغذائي الخلقي، وهو من أوائل الأمراض التي تم اكتشافها.
بيلة الفينيل كيتون - نوع الميراث
مرض فيلينغ هو مرض وراثي كروموسومي ينتقل إلى الأطفال من الوالدين. السبب وراء تطور علم الأمراض هو الجين الموجود على الكروموسوم 12. وهو مسؤول عن إنتاج إنزيم الكبد فينيل ألانين 4-هيدروكسيلاز، الذي يحول الفينيل ألانين إلى مادة أخرى - التيروزين (وهو ضروري لأداء الجسم الطبيعي).
لقد ثبت أن بيلة الفينيل كيتون موروثة كصفة متنحية. يحمل حوالي 2% من الأشخاص الجين المعيب ولكن لا يعانون من بيلة الفينيل كيتون. يتطور علم الأمراض فقط عندما ينقل كل من الأم والأب الجين إلى الطفل، ويمكن أن يحدث هذا باحتمال 25٪. إذا تم توريث بيلة الفينيل كيتون كصفة متنحية، وكانت الزوجة متغايرة الزيجوت، والزوج متماثل الزيجوت بالنسبة للأليل الطبيعي للجين، فإن احتمال أن يكون الأطفال حاملين أصحاء لجين بيلة الفينيل كيتون هو 50٪.
أشكال بيلة الفينيل كيتون
عند النظر في الأشخاص الذين يمكن أن يصابوا ببيلة الفينيل كيتون ونوع المرض، فإننا غالبًا ما نتحدث عن الشكل الكلاسيكي للمرض، والذي يحدث في حوالي 98٪ من الحالات. الحالات المتبقية هي العامل المساعد فينيل كيتون يوريا، الناجم عن خلل في رباعي هيدروبيوبترين بسبب انتهاك تخليقه أو استعادة الشكل النشط. تعمل هذه المادة كعامل مساعد لعدد من الإنزيمات، وبدونها يكون نشاطها مستحيلا.

بيلة الفينيل كيتون - الأسباب
مرض فيلينغ هو مرض يحدث فيه، بسبب الطفرات في الجين الذي يسبب نقص أو غياب الفينيل ألانين 4-هيدروكسيلاز، تراكم الفينيل ألانين، وكذلك منتجات انهياره غير الكامل، في الأنسجة والسوائل الفسيولوجية. ويتحول بعض من الفينيل ألانين الزائد إلى فينيل كيتونات، والتي تفرز في البول، ومن هنا جاء اسم المرض.
يؤثر انتهاك عمليات التمثيل الغذائي على الدماغ إلى حد أكبر. يتم إنتاج تأثير سام على أنسجتها، وتتعطل عمليات التمثيل الغذائي للدهون، وتفشل عملية تكوين الميالين للألياف العصبية، ويتم تقليل تكوين الناقلات العصبية. هكذا يبدأ إطلاق الآليات المرضية للتخلف العقلي لدى الطفل.
بيلة الفينيل كيتون - الأعراض
عند الولادة، يبدو الطفل المصاب بهذا التشخيص بصحة جيدة، وفقط بعد 2-6 أشهر تظهر الأعراض الأولى. تبدأ أعراض بيلة الفينيل كيتون في إظهار علامات عندما يتراكم الفينيل ألانين، الذي يأتي مع حليب الثدي أو الرضاعة الصناعية، في جسم الطفل. قد تحدث الأعراض التالية، غير المحددة بعد:
- الخمول المفرط أو الأرق.
- صرخات بلا سبب.
- قلس متكرر.
- اضطرابات النوم.
بالإضافة إلى ذلك، يتمتع الأطفال المرضى ببشرة وشعر وعينين أفتح من أفراد الأسرة الأصحاء، وهو ما يرتبط بخلل في إنتاج صبغة الميلانين في الجسم. علامة تشخيصية أخرى قد يلاحظها الأطباء أو الآباء اليقظون هي رائحة "الفأر" الغريبة الناتجة عن إطلاق مادة الفينيللانين في البول والعرق.
تصبح المظاهر السريرية أكثر وضوحًا عند عمر ستة أشهر تقريبًا، بعد إدخال الأطعمة التكميلية الأولى:
- عدم القدرة على التركيز على الأشياء الفردية.
- اللامبالاة بكل ما يحدث؛
- قلة تعابير الوجه والابتسامات.
- هزات اليد وما إلى ذلك.
كما يمكن ملاحظة التشوهات الجسدية: صغر حجم الرأس، وبروز الفك العلوي، وتوقف النمو. يبدأ الأطفال المرضى في رفع رؤوسهم، والزحف، والجلوس، والوقوف في وقت متأخر. تتميز وضعية الجلوس الخاصة - وضعية "الخياط" حيث يتم ثني الذراعين باستمرار عند المرفقين والساقين عند الركبتين. وفي سن ثلاث سنوات، إذا لم يبدأ العلاج، تزداد الأعراض.

بيلة الفينيل كيتون - التشخيص
غالبًا ما يتم اكتشاف بيلة الفينيل كيتون عند الأطفال في مستشفى الولادة، مما يجعل من الممكن بدء العلاج في الوقت المحدد ومنع تطور عدد من العواقب التي لا رجعة فيها. في الأيام 4-5 بعد الولادة، يتم أخذ الدم الشعري من الأطفال على معدة فارغة لتحديد بعض الأمراض الوراثية الشديدة، بما في ذلك بيلة الفينيل كيتون. إذا حدث الخروج من مستشفى الولادة في وقت سابق، يتم إجراء التحليل في العيادة في مكان الإقامة خلال الأيام العشرة الأولى من الحياة.
ونظرًا لأنه في حالات نادرة تكون هناك نتائج خاطئة، فلا يتم التشخيص أبدًا بناءً على نتائج التحليل الأول. لتأكيد علم الأمراض الحالي، يتم وصف عدد من الدراسات الأخرى، بما في ذلك:
- اختبار البول للكشف عن فينيل بيروفات.
- التحديد الكمي للفينيل ألانين والتيروزين في البلازما؛
- تحديد نشاط انزيم الكبد.
- تخطيط كهربية الدماغ والتصوير بالرنين المغناطيسي للدماغ.
يمكن اكتشاف خلل وراثي يؤدي إلى تطور الأمراض في الجنين أثناء التشخيص الغزوي قبل الولادة. وللقيام بذلك، يتم أخذ عينات من الخلايا من الزغب المشيمي أو السائل الأمنيوسي، ومن ثم يتم إجراء تحليل الحمض النووي. يوصى بإجراء مثل هذه الدراسة في العائلات ذات الخطورة العالية للإصابة بالأمراض، بما في ذلك إذا كان هناك بالفعل طفل مصاب ببيلة الفينيل كيتون.
بيلة الفينيل كيتون - العلاج
عندما يتم اكتشاف بيلة الفينيل كيتون عند الأطفال حديثي الولادة، يجب مراقبة المرضى من قبل أطباء من تخصصات مثل اختصاصي الوراثة، وطبيب الأطفال، وطبيب الأعصاب، وأخصائي التغذية. بالنسبة لأولئك الذين يعرفون ما هو نوع مرض بيلة الفينيل كيتون، سيكون من الواضح لماذا أساس علاجه هو اتباع نظام غذائي محدود في الفينيل ألانين. بالإضافة إلى ذلك، يوصف العلاج الدوائي والتدليك والعلاج الطبيعي والأساليب النفسية والتربوية لتنشئة الطفل اجتماعيًا وإعداده للتعليم.

عندما يتم تشخيص بيلة الفينيل كيتون، يتم وصف نظام غذائي للطفل على الفور. يتم استبعاد الأطعمة الغنية بالبروتين (اللحوم والأسماك ومنتجات الألبان والبقوليات والمكسرات وغيرها) من النظام الغذائي. يتم تعويض الحاجة إلى البروتينات عن طريق مخاليط غذائية خاصة ومنتجات أخرى تحتوي على بيرلوفين، وهو بروتين هيدروليزات شبه اصطناعي خالٍ تمامًا من الفينيل ألانين (Tetrafen، Lofenalac، Nophelan). يأكل المرضى الخبز الخالي من البروتين والمعكرونة والحبوب والموس وما إلى ذلك. تتم الرضاعة الطبيعية بجرعات محدودة.
إن الالتزام الصارم بنظام غذائي مع مراقبة منتظمة لمستويات الفينيل ألانين في الدم خلال السنوات 14-15 الأولى من الحياة يمنع تطور الاضطرابات العقلية. علاوة على ذلك، يتوسع النظام الغذائي إلى حد ما، لكن العديد من الخبراء يوصون بالالتزام بنظام غذائي خاص مدى الحياة. لا يمكن علاج العامل المساعد لبيلة الفينيل كيتون من خلال النظام الغذائي، ولكن يتم تصحيحه فقط عن طريق إعطاء مستحضرات رباعي هيدروبيوبترين.







