La distrofia è un processo patologico che è una conseguenza di una violazione dei processi metabolici, con danni alle strutture cellulari e comparsa di sostanze nelle cellule e nei tessuti del corpo che normalmente non vengono rilevate.
Le distrofie sono classificate:
1) dalla scala della prevalenza del processo: locale (localizzato) e generale (generalizzato);
2) a causa dell'occorrenza: acquisita e congenita. Le distrofie congenite hanno una condizione genetica della malattia.
Le distrofie ereditarie si sviluppano a causa di una violazione del metabolismo di proteine, carboidrati, grassi, in questo caso è importante la carenza genetica di uno o un altro enzima coinvolto nel metabolismo di proteine, grassi o carboidrati. Successivamente nei tessuti si accumulano prodotti non completamente convertiti del metabolismo dei carboidrati, delle proteine e dei grassi. Questo processo può svilupparsi in vari tessuti del corpo, ma il tessuto del sistema nervoso centrale è necessariamente danneggiato. Tali malattie sono chiamate malattie da accumulo. I bambini affetti da queste malattie muoiono nel 1° anno di vita. Maggiore è la mancanza dell'enzima necessario, più rapido è lo sviluppo della malattia e prima si verifica la morte.
Le distrofie si dividono in:
1) in base al tipo di metabolismo disturbato: proteine, carboidrati, grassi, minerali, acqua, ecc.;
2) secondo il punto di applicazione (secondo la localizzazione del processo): cellulari (parenchima), non cellulari (mesenchimale), che si sviluppano nel tessuto connettivo, nonché misti (osservati sia nel parenchima che nel tessuto connettivo).
I meccanismi patogenetici sono quattro.
1. Trasformazione- è la capacità di alcune sostanze di trasformarsi in altre di struttura e composizione simile. Ad esempio, i carboidrati hanno questa capacità, trasformandosi in grassi.
2. Infiltrazione- questa è la capacità delle cellule o dei tessuti di riempirsi di una quantità eccessiva di varie sostanze. Esistono due tipi di infiltrazione. Per l'infiltrazione del primo tipo è caratteristico che una cellula che partecipa alla vita normale riceva una quantità eccessiva di sostanza. Dopo un po ', arriva un limite in cui la cellula non può elaborare e assimilare questo eccesso. L'infiltrazione del secondo tipo è caratterizzata da una diminuzione del livello di attività vitale della cellula, di conseguenza non riesce nemmeno a far fronte alla normale quantità di sostanza che vi entra.
3. Decomposizione- caratterizzato dalla disintegrazione delle strutture intracellulari e interstiziali. C'è una rottura dei complessi proteici-lipidici che fanno parte delle membrane degli organelli. Nella membrana proteine e lipidi sono in uno stato legato e quindi non sono visibili. Ma quando le membrane si rompono, si formano nelle cellule e diventano visibili al microscopio.
4. Sintesi perversa- Nella cellula si formano sostanze estranee anomale che non si formano durante il normale funzionamento del corpo. Ad esempio, nella degenerazione amiloide, le cellule sintetizzano una proteina anomala, dalla quale si forma poi l’amiloide. Nei pazienti con alcolismo cronico nelle cellule del fegato (epatociti), inizia a verificarsi la sintesi di proteine estranee, dalle quali si forma successivamente la cosiddetta ialina alcolica.
Diversi tipi di distrofie sono caratterizzati dalla disfunzione del tessuto. Nella distrofia, il disturbo è duplice: quantitativo, con una diminuzione della funzione, e qualitativo, con una perversione della funzione, cioè compaiono caratteristiche che non sono caratteristiche di una cellula normale. Un esempio di tale funzione perversa è la comparsa di proteine nelle urine nelle malattie renali, quando si verificano cambiamenti distrofici nei reni o cambiamenti nei test epatici che compaiono nelle malattie del fegato e nelle malattie cardiache - un cambiamento nel tono cardiaco.
Le distrofie parenchimali si dividono in proteine, grassi e carboidrati.
Distrofia proteica- Questa è una distrofia in cui il metabolismo delle proteine è disturbato. Il processo di distrofia si sviluppa all'interno della cellula. Tra le distrofie del parenchima proteico si distinguono le distrofie granulari, a goccia ialina e idropiche.
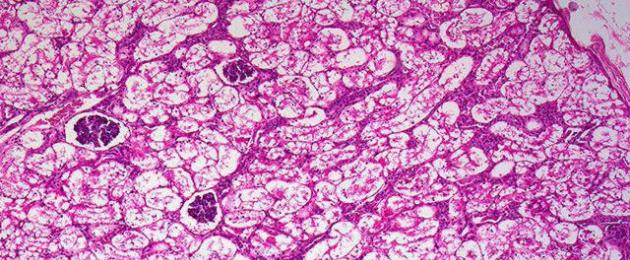
Con la distrofia granulare, durante l'esame istologico, si possono vedere grani proteici nel citoplasma delle cellule. La distrofia granulare colpisce gli organi parenchimali: reni, fegato e cuore. Questa distrofia è chiamata gonfiore torbido o opaco. Ciò è legato alle caratteristiche macroscopiche. Gli organi affetti da questa distrofia si gonfiano leggermente e la superficie del taglio appare opaca, torbida, come se fosse “scottata con acqua bollente”.
Contribuisce allo sviluppo della distrofia granulare per diversi motivi, che possono essere suddivisi in 2 gruppi: infezioni e intossicazioni. Un rene affetto da distrofia granulare aumenta di dimensioni, diventa flaccido, si può determinare un test di Schorr positivo (quando i poli del rene vengono uniti, il tessuto renale viene strappato). Nella sezione, il tessuto è opaco, i confini del midollo e della corteccia sono sfumati o possono essere del tutto indistinguibili. Con questo tipo di distrofia viene colpito l'epitelio dei tubuli contorti del rene. Nei normali tubuli renali si osservano anche degli spazi vuoti e nella distrofia granulare il citoplasma apicale viene distrutto e il lume diventa a forma di stella. Nel citoplasma dell'epitelio dei tubuli renali sono presenti numerosi granuli (rosa).
La distrofia granulare renale termina in due varianti. Un risultato favorevole è possibile quando la causa viene eliminata, l'epitelio dei tubuli in questo caso ritorna alla normalità. Un esito sfavorevole si verifica con l'esposizione continua a un fattore patologico: il processo diventa irreversibile, la distrofia si trasforma in necrosi (spesso osservata in caso di avvelenamento con veleni renali).
Anche il fegato con distrofia granulare è leggermente ingrossato. Una volta tagliato, il tessuto acquisisce il colore dell'argilla. Il segno istologico della degenerazione granulare del fegato è la presenza incoerente di granuli proteici. È necessario prestare attenzione se la struttura della trave è presente o distrutta. Con questa distrofia, le proteine sono divise in gruppi localizzati separatamente o epatociti che giacciono separatamente, che è chiamata discomplessazione dei fasci epatici.
Distrofia granulare cardiaca: anche il cuore è leggermente ingrossato verso l'esterno, il miocardio diventa flaccido, al taglio ricorda la carne bollita. Macroscopicamente non si osservano granuli proteici.
Nell'esame istologico, il criterio per questa distrofia è la basofilia. Le fibre miocardiche percepiscono diversamente l'ematossilina e l'eosina. Alcune aree delle fibre sono intensamente colorate con ematossilina in lilla, mentre altre sono intensamente colorate con eosina in blu.
La degenerazione delle goccioline ialine si sviluppa nei reni (è interessato l'epitelio dei tubuli contorti). Si verifica in malattie renali come la glomerulonefrite cronica, la pielonefrite cronica e in caso di avvelenamento. Gocce di una sostanza simile alla ialina si formano nel citoplasma delle cellule. Tale distrofia è caratterizzata da una significativa violazione della filtrazione renale.
La distrofia idropica può verificarsi nelle cellule del fegato nell'epatite virale. Allo stesso tempo, negli epatociti si formano grandi gocce luminose, che spesso riempiono la cellula.
Degenerazione grassa. Esistono 2 tipi di grassi. La quantità di grassi mobili (labili) cambia nel corso della vita di una persona, sono localizzati nei depositi di grasso. I grassi stabili (fissi) sono inclusi nella composizione delle strutture cellulari, delle membrane.
I grassi svolgono un'ampia varietà di funzioni: di supporto, protettive, ecc.
I grassi vengono determinati utilizzando coloranti speciali:
1) Sudan-III ha la capacità di colorare il grasso in rosso-arancio;
2) colori scarlatti rosso;
3) il Sudan-IV (acido osmico) si colora di nero grasso;
4) Il blu Nilo ha metacromasia: colora di rosso i grassi neutri e tutti gli altri grassi diventano blu o blu sotto la sua influenza.
Immediatamente prima della colorazione, il materiale originale viene lavorato utilizzando due metodi: il primo è il cablaggio con alcool, il secondo è il congelamento. Per la determinazione dei grassi viene utilizzato il congelamento delle sezioni di tessuto, poiché i grassi si dissolvono negli alcoli.
I disturbi del metabolismo dei grassi sono tre patologie:
1) vera e propria degenerazione grassa (cellulare, parenchimale);
2) obesità generale o obesità;
3) obesità della sostanza interstiziale delle pareti dei vasi sanguigni (aorta e suoi rami).
In realtà la degenerazione grassa è alla base dell'aterosclerosi. Le cause della degenerazione grassa possono essere suddivise in due gruppi principali: infezioni e intossicazioni. Al giorno d'oggi, il principale tipo di intossicazione cronica è l'intossicazione da alcol. Spesso può verificarsi intossicazione da farmaci, intossicazione endocrina, che si sviluppa nel diabete mellito.
Un esempio di infezione che provoca degenerazione grassa è la difterite, poiché la tossina difterica può causare la degenerazione grassa del miocardio. La degenerazione grassa si osserva negli stessi organi delle proteine: nel fegato, nei reni e nel miocardio.
Con la degenerazione grassa, il fegato aumenta di dimensioni, diventa denso, al taglio è opaco, di colore giallo brillante. Questo tipo di fegato ha ricevuto il nome figurato "fegato d'oca".
Manifestazioni microscopiche: nel citoplasma degli epatociti compaiono gocce di grasso di piccole, medie e grandi dimensioni. Di norma, si trovano al centro del lobulo epatico, ma possono occuparlo interamente.
Ci sono diverse fasi nel processo di obesità:
1) obesità semplice, quando una goccia occupa l'intero epatocita, ma quando cessa l'impatto del fattore patologico (quando il paziente smette di bere alcolici), dopo 2 settimane il fegato ritorna normale;
2) necrosi: si verifica un'infiltrazione di leucociti attorno al fuoco della necrosi come risposta al danno; il processo in questa fase è reversibile;
3) fibrosi - cicatrici; il processo entra in uno stadio cirrotico irreversibile.
C'è un ingrossamento del cuore, il muscolo diventa flaccido, opaco e, se esamini attentamente l'endocardio, sotto l'endocardio dei muscoli papillari, puoi vedere una striatura trasversale, chiamata "cuore della tigre".
Caratteristiche microscopiche: il grasso è presente nel citoplasma dei cardiomiociti. Il processo ha un carattere a mosaico: la lesione patologica si estende ai cardiomiociti situati lungo le piccole vene. L'esito può essere favorevole quando si verifica un ritorno alla normalità (se la causa viene eliminata) e se la causa continua ad agire, si verifica la morte cellulare e al suo posto si forma una cicatrice.
Nei reni il grasso è localizzato nell'epitelio dei tubuli contorti. Tale distrofia si verifica nelle malattie renali croniche (nefrite, amiloidosi), in caso di avvelenamento, obesità generale.
L'obesità sconvolge il metabolismo dei grassi labili neutri, che si formano in eccesso nei depositi di grasso; il peso corporeo aumenta in modo significativo a causa dell'accumulo di grasso nel tessuto adiposo sottocutaneo, nell'omento, nel mesentere, nel tessuto perirenale, retroperitoneale, nel tessuto che ricopre il cuore. Con l'obesità, il cuore diventa, per così dire, intasato da una spessa massa grassa, e quindi il grasso penetra nello spessore del miocardio, provocandone la degenerazione grassa. Le fibre muscolari subiscono la pressione dello stroma grasso e dell'atrofia, che porta allo sviluppo di insufficienza cardiaca. Molto spesso, viene interessato lo spessore del ventricolo destro, a seguito del quale si sviluppa una congestione nella circolazione sistemica. Inoltre, l’obesità del cuore può provocare la rottura del miocardio. Nelle fonti letterarie, un cuore così grasso è caratterizzato dalla sindrome di Pickwick.
Nel fegato con obesità si può formare grasso all’interno delle cellule. Il fegato assume l'aspetto di un "fegato d'oca", come nella distrofia. È possibile differenziare il grasso risultante nelle cellule del fegato mediante colorazione: il blu Nilo ha la capacità di colorare il grasso neutro in rosso nell'obesità e in blu nella distrofia avanzata.
Obesità della sostanza interstiziale delle pareti dei vasi sanguigni (ovvero lo scambio di colesterolo): durante l'infiltrazione dal plasma sanguigno nella parete vascolare già preparata, entra il colesterolo, che si deposita poi sulla parete vascolare. Una parte viene lavata indietro e una parte viene elaborata dai macrofagi. I macrofagi carichi di grasso sono chiamati cellule di xantoma. Sopra i depositi di grasso cresce il tessuto connettivo, che sporge nel lume della nave, formando così una placca aterosclerotica.
Cause dell'obesità:
1) geneticamente determinato;
2) endocrino (diabete, malattia di Itsenko-Cushing);
3) ipodynamia;
4) eccesso di cibo.
degenerazione dei carboidrati può essere associato ad un alterato metabolismo del glicogeno o delle glicoproteine. La violazione del contenuto di glicogeno si manifesta con una diminuzione o un aumento della sua quantità nei tessuti e con l'aspetto in cui di solito non viene rilevato. Questi disturbi sono espressi nel diabete mellito e nelle distrofie ereditarie dei carboidrati - glicogenosi.
Nel diabete mellito si verifica un consumo insufficiente di glucosio da parte dei tessuti, un aumento della sua quantità nel sangue (iperglicemia) e l'escrezione nelle urine (glicosuria). Le riserve di glicogeno nei tessuti vengono drasticamente ridotte. Nel fegato si verifica una violazione della sintesi del glicogeno, che porta all'infiltrazione dei suoi grassi: si verifica la degenerazione grassa del fegato. Allo stesso tempo, nei nuclei degli epatociti compaiono inclusioni di glicogeno, che diventano leggeri (nuclei "perforati" e "vuoti"). Con la glicosuria compaiono cambiamenti nei reni, manifestati nell'infiltrazione di glicogeno nell'epitelio dei tubuli. L'epitelio diventa alto, con citoplasma leggero e schiumoso; granuli di glicogeno si trovano anche nel lume dei tubuli. I tubuli renali diventano più permeabili alle proteine plasmatiche e agli zuccheri. Si sviluppa una delle manifestazioni della microangiopatia diabetica: la glomerulosclerosi intercapillare (diabetica). La glicogenosi è causata dall'assenza o dall'insufficienza di un enzima coinvolto nella degradazione del glicogeno immagazzinato e si riferisce alle fermentopatie ereditarie (malattie da accumulo).
Con le distrofie dei carboidrati associate a una violazione del metabolismo delle glicoproteine, si verifica un accumulo di mucine e mucoidi, chiamate anche sostanze mucose e simili al muco (degenerazione della mucosa). Le cause sono varie, ma il più delle volte si tratta di un'infiammazione delle mucose. La distrofia sistemica è alla base di una malattia sistemica ereditaria: la fibrosi cistica. Sono colpiti l'apparato endocrino del pancreas, le ghiandole dell'albero bronchiale, l'apparato digerente e urinario, le vie biliari, le ghiandole genitali e le mucose. Il risultato è diverso: in alcuni casi si verifica la rigenerazione dell'epitelio e il completo ripristino della mucosa, mentre in altri si atrofizza, si sclerosi e la funzione dell'organo è compromessa.
La distrofia stroma-vascolare è un disturbo metabolico del tessuto connettivo, principalmente nella sua sostanza intercellulare, l'accumulo di prodotti metabolici. A seconda del tipo di metabolismo compromesso, le distrofie mesenchimali sono suddivise in proteine (disproteinosi), grassi (lipidosi) e carboidrati. Tra le disproteinosi si distinguono gonfiore mucoide, gonfiore fibrinoso, ialinosi e amiloidosi. I primi tre sono associati a una violazione della permeabilità della parete vascolare.
1. Gonfiore mucoideè un processo reversibile. Ci sono cambiamenti superficiali superficiali nella struttura del tessuto connettivo. A causa dell'azione di un fattore patologico, nella sostanza principale si verificano processi di decomposizione, cioè i legami delle proteine e degli aminoglicani si rompono. Gli aminoglicani sono liberi e si trovano nel tessuto connettivo. A causa loro, il tessuto connettivo si colora in modo basofilo. Esiste un fenomeno di metacromasia (la capacità del tessuto di cambiare il colore della tintura). Quindi, il blu di toluidina è normalmente blu e con gonfiore mucoide è rosa o lilla. La mucina (muco) è costituita da proteine e quindi si colora in modo peculiare. I glucosaminoglicani assorbono bene il fluido che esce dal letto vascolare e le fibre si gonfiano, ma non collassano. Il quadro macroscopico non è cambiato. I fattori che causano il gonfiore del muco includono: ipossia (ipertensione, aterosclerosi), disturbi immunitari (malattie reumatiche, disturbi endocrini, malattie infettive).
2. gonfiore fibrinoide- si tratta di una disorganizzazione profonda e irreversibile del tessuto connettivo, che si basa sulla distruzione della sostanza base del tessuto e delle fibre, accompagnata da un forte aumento della permeabilità vascolare e dalla formazione di fibrinoidi. Potrebbe essere dovuto al gonfiore del muco. Le fibre vengono distrutte, il processo è irreversibile. La proprietà della metacromasia scompare. Il quadro macroscopico è invariato. Fibre di collagene osservate al microscopio impregnate di proteine plasmatiche, colorate in giallo con pirofucsina.
L'esito del gonfiore fibrinoide può essere necrosi, ialinosi, sclerosi. Intorno alla zona di gonfiore fibrinoide si accumulano macrofagi, sotto l'influenza dei quali le cellule vengono distrutte e si verifica la necrosi. I macrofagi sono in grado di produrre monochine, che promuovono la riproduzione dei fibroblasti. Pertanto, l'area di necrosi viene sostituita dal tessuto connettivo: si verifica la sclerosi.
3. Distrofia ialina (ialinosi). Nel tessuto connettivo si formano masse omogenee trasparenti e dense di ialina (proteina fibrillare), resistenti agli alcali, agli acidi, agli enzimi, PAS-positivi, coloranti acidi ben percepiti (eosina, fucsina acida), colorati di giallo o rosso con pirofucsina.
La ialinosi è il risultato di vari processi: infiammazione, sclerosi, gonfiore dei fibrinoidi, necrosi, impregnazione del plasma. Distinguere tra ialinosi dei vasi e tessuto connettivo stesso. Ciascuno può essere diffuso (sistemico) e locale.
Con la ialinosi dei vasi, sono colpite principalmente le piccole arterie e le arteriole. Microscopicamente: la ialina si trova nello spazio subendoteliale, distruggendo la placca elastica, la nave si trasforma in un tubo vitreo ispessito con un lume molto ristretto o completamente chiuso.
La ialinosi dei piccoli vasi è sistemica, ma espressa in modo significativo nei reni, nel cervello, nella retina, nel pancreas. Caratteristico per l'ipertensione, la microangiopatia diabetica e le malattie con compromissione dell'immunità.
Esistono tre tipi di ialine vascolari:
1) semplice, derivante dall'assorbimento di componenti del plasma sanguigno invariati o leggermente modificati (con ipertensione, aterosclerosi);
2) lipogialina contenente lipidi e α-lipoproteine (nel diabete mellito);
3) complesso ialino, costituito da complessi immunitari, strutture collassanti della parete vascolare, fibrina (tipico per malattie con disturbi immunopatologici - ad esempio per malattie reumatiche).
La ialinosi del tessuto connettivo stesso si sviluppa a causa del rigonfiamento dei fibrinoidi, che porta alla distruzione del collagene e all'impregnazione del tessuto con proteine plasmatiche e polisaccaridi. L'aspetto dell'organo cambia, si verifica la sua atrofia, si verificano deformazioni e rughe. Il tessuto connettivo diventa denso, biancastro e traslucido. Microscopicamente: il tessuto connettivo perde la fibrillazione e si fonde in una massa omogenea densa simile alla cartilagine; gli elementi cellulari vengono compressi e vanno incontro ad atrofia.
Nella ialinosi locale l'esito sono cicatrici, aderenze fibrose di cavità sierose, sclerosi vascolare, ecc. Nella maggior parte dei casi l'esito è sfavorevole, ma è anche possibile il riassorbimento delle masse ialine.
4. Amiloidosi- una sorta di distrofia proteica, che è una complicazione di varie malattie (di natura infettiva, infiammatoria o tumorale). In questo caso si tratta di un'amiloidosi acquisita (secondaria). Quando l’amiloidosi deriva da un’eziologia sconosciuta, si parla di amiloidosi primaria. La malattia è stata descritta da K. Rakitansky ed è stata chiamata "malattia sebacea", poiché il segno microscopico dell'amiloidosi è la lucentezza sebacea dell'organo. L'amiloide è una sostanza complessa - una glicoproteina, in cui le proteine globulari e fibrillari sono strettamente correlate ai mucopolisaccaridi. Se le proteine hanno approssimativamente la stessa composizione, i polisaccaridi hanno sempre una composizione diversa. Di conseguenza, l’amiloide non ha mai una composizione chimica costante. La percentuale di proteine è pari al 96-98% della massa totale di amiloide. Esistono due frazioni di carboidrati: polisaccaridi acidi e neutri. Le proprietà fisiche dell'amiloide sono rappresentate dall'anisotropia (la capacità di birifrangenza, che si manifesta nella luce polarizzata), al microscopio l'amiloide produce un bagliore giallo, che differisce dal collagene e dall'elastina. Reazioni colorate per la determinazione dell'amiloide: la colorazione elettiva "Rosso Congo" colora l'amiloide di un colore rosso mattone, dovuto alla presenza di fibrille nella composizione dell'amiloide, che hanno la capacità di legare e trattenere saldamente il colorante .
Reazioni metacromatiche: verde iodio, viola di metile, viola di genziana si colorano di rosso amiloide su sfondo verde o blu. La colorazione avviene a causa dei glicosaminoglicani. La tecnica più sensibile è il trattamento con fluorocromo (tioflavina S, F). Con questo metodo è possibile rilevare depositi minimi di amiloide. Potrebbe esserci amiloide acromatica che non si colora completamente; in questo caso viene utilizzata la microscopia elettronica. Al microscopio elettronico diventano visibili 2 componenti: la componente F - fibrille e la componente P - bastoncelli periodici. Le fibrille sono due fili paralleli, i bastoncini periodici sono costituiti da formazioni pentagonali.
Assegni il collegamento IV di morfogenesi.
I. Trasformazione cellulare del sistema reticoloendoteliale, che precede la formazione di cloni cellulari - amiloidoblasti.
II Sintesi da parte degli amiloidoblasti del componente principale della proteina amiloide-fibrillare.
III Aggregazione di fibrille tra loro con formazione di una struttura amiloide.
IV. La connessione delle fibrille aggregate con le proteine del plasma sanguigno, nonché con i glicosaminoglicani tissutali, che porta alla precipitazione di una sostanza anormale, l'amiloide, nei tessuti.
Nella prima fase, la formazione di plasmacellule negli organi del sistema reticoloendoteliale (plasmatizzazione del midollo osseo, milza, linfonodi, fegato). La plasmatizzazione si nota anche nello stroma degli organi. Le plasmacellule si sviluppano in cellule amiloidi. La sintesi delle proteine fibrillari avviene sempre nelle cellule di origine mesenchimale. Si tratta di linfociti, plasmacellule, fibroblasti, cellule reticolari (i fibroblasti si trovano più spesso nell'amiloidosi familiare), plasmacellule nell'amiloidosi primaria (causata da un tumore), cellule reticolari nell'amiloidosi secondaria. Inoltre, le cellule di Kupffer del fegato, gli endoteliociti stellati, le cellule mesangiali (nel rene) possono agire come amiloidoblasti. Quando la proteina si accumula abbastanza, si forma un'impalcatura.
La proteina fibrillare è considerata estranea, anormale. In risposta alla sua formazione appare un ulteriore gruppo di cellule che inizia a tentare di lisare l'amiloide. Queste cellule sono chiamate amiloidoclasti. La funzione di tali cellule può essere eseguita da macrofagi liberi e fissi. Per molto tempo si svolge una lotta alla pari tra le cellule che formano e dissolvono l'amiloide, ma finisce sempre con la vittoria degli amiloidoblasti, poiché nei tessuti si verifica la tolleranza immunologica alla proteina delle fibrille amiloidi. Sullo scheletro fibrillare si depositano proteine e polisaccaridi.
L'amiloide si forma sempre all'esterno delle cellule e ha sempre una stretta connessione con le fibre del tessuto connettivo: con le fibre reticolari e di collagene. Se la perdita di amiloide avviene lungo le fibre reticolari nelle membrane dei vasi sanguigni o delle ghiandole, allora si chiama amiloide perireticolare (parenchimale) e si osserva nella milza, nel fegato, nei reni, nelle ghiandole surrenali e nell'intestino. Se la formazione e la perdita di amiloide ricade sulle fibre di collagene, allora viene chiamata pericollagene o mesenchimale. In questo caso vengono colpiti l'avventizia dei grandi vasi, lo stroma miocardico, i muscoli striati e lisci, i nervi e la pelle.
Esistono 3 vecchie teorie e 1 nuova teoria moderna che combina tutte e tre le teorie sulla patogenesi dell'amiloidosi.
1. Teoria della disproteinosi. Secondo questa teoria, si sviluppa la disproteinemia, con essa si verifica un accumulo nel plasma sanguigno di frazioni proteiche grossolane e proteine anormali - paraproteine. Appaiono a causa del metabolismo proteico compromesso. Quindi vanno oltre il letto vascolare, interagiscono con i mucopolisaccaridi tissutali. Questa teoria è semplice e non spiega la comparsa della disproteinemia.
2. teoria immunologica. In varie malattie, nel sangue si accumulano prodotti di decomposizione dei tessuti, i leucociti, anche le tossine batteriche circolano: tutte queste sostanze hanno proprietà antigeniche e portano alla formazione di anticorpi contro se stesse. Si sviluppa una reazione immunitaria per combinare gli antigeni con gli anticorpi nei luoghi in cui sono stati prodotti gli anticorpi, cioè negli organi del sistema reticoloendoteliale. Questa teoria spiega solo una parte della degenerazione amiloide, cioè dove c'è suppurazione cronica, e non spiega le forme genetiche dell'amiloidosi.
3. Teoria della sintesi cellulare-locale. Questa teoria studia l'amiloide come un segreto delle cellule mesenchimali.
4. teoria universale- mutazionale. I fattori mutageni colpiscono le cellule provocando mutazioni e si innesca un meccanismo che porta alla formazione di cellule amiloidoblastiche.
Esistono forme secondarie, o acquisite, e idiopatiche (primarie), ereditarie (familiari, senili, tumorali). La forma secondaria è una complicazione di un'ampia varietà di infezioni. Le cause dell’amiloidosi primaria sono sconosciute.
Le amiloidosi secondarie sono localizzate perireticolarmente, hanno un effetto devastante sugli organi parenchimali. Gli amiloidi secondari cadono lungo il percorso delle fibre di collagene. Molto spesso si verificano lesioni di origine mesenchimale. Nella forma idiopatica sono colpiti il cuore, i nervi e l’intestino. Con l'amiloidosi ereditaria o familiare, c'è un effetto sui gangli nervosi simpatici e sugli organi parenchimali: i reni. Caratteristica è la cosiddetta malattia periodica, osservata in persone delle nazionalità più antiche, ad esempio ebrei, arabi, armeni. Nella forma senile sono colpiti il cuore e le vescicole seminali.
L'amiloidosi tumorale è così chiamata perché la deposizione di amiloide che si verifica con essa assomiglia a un tumore. Colpisce le vie respiratorie, la trachea, la vescica, la pelle, la congiuntiva.
Le cause dell’amiloidosi secondaria includono:
1) malattie polmonari croniche aspecifiche, come bronchite cronica con bronchiectasie, ascessi polmonari cronici, bronchiectasie;
2) tubercolosi in forma cavernosa;
3) artrite reumatoide (circa il 25%).
Caratteristiche macroscopiche: gli organi sono ingranditi, densi, fragili, si rompono facilmente, il bordo dell'incisione è affilato, poiché l'amiloide si deposita sotto la membrana vascolare, causandone il restringimento, si sviluppa l'ischemia e l'organo diventa pallido. L'amiloide conferisce al corpo una caratteristica lucentezza grassa.
Durante l'autopsia degli organi viene utilizzato un test macroscopico di Virchow per l'amiloide. Il test viene eseguito su organi freschi, non fissati: una piastra viene prelevata dall'organo, lavata con acqua di sangue e annaffiata con la soluzione di Lugol, e dopo 30 minuti l'organo viene annaffiato con acido solforico al 10%. Quando appare una macchia sulla bottiglia sporca, il test è positivo.
La milza è colpita nello stadio II. Nella prima fase, l'amiloide si accumula nei follicoli della milza, nella polpa bianca, e assomiglia a granelli bianchi. Sembrano grani di sago e una tale milza si chiama sago. Nella seconda fase, l’amiloide si diffonde in tutto l’organo. La milza aumenta notevolmente di dimensioni, consistenza densa, rosso-brunastro con una lucentezza untuosa sul taglio. Ha ricevuto il nome di milza grassa (prosciutto).
Nel rene, l'amiloide appare sotto la membrana dei capillari glomerulari, sotto la membrana dei vasi del midollo e dello strato corticale, sotto le membrane dei tubuli contorti e diritti, e anche nello stroma del rene lungo le fibre reticolari. Questo processo è costante: il primo stadio: l'amiloide latente (latente) inizia a formarsi nelle piramidi, nei vasi sanguigni glomerulari; il secondo stadio è caratterizzato da proteinuria. Una grande quantità di proteine viene determinata nelle urine. Nello stroma si notano fenomeni di sclerosi, dovuti allo sviluppo di ischemia. Nell'epitelio si trovano segni di distrofia grassa e a goccia ialina.
Il terzo stadio è nefrotico. I cambiamenti macroscopici corrispondono a un grande rene sebaceo: l'organo è significativamente ingrandito, uno strato corticale spesso e piuttosto pallido con una lucentezza grassa e piramidi viola-bluastre gonfie. L'immagine al microscopio mostra che tutti i glomeruli contengono amiloide diffusamente localizzata. L'ultimo stadio finale è uremico. In questa fase si sviluppano le rughe dei reni. L'insufficienza renale porta alla morte.
Nel fegato inizia la deposizione di amiloide nei sinusoidi tra le cellule di Kupffer, lungo lo stroma reticolare dei lobuli, le cellule del fegato vengono compresse e muoiono per atrofia. Nelle ghiandole surrenali, l'amiloide si deposita solo nello strato corticale lungo i capillari, il che porta all'insufficienza surrenalica, quindi qualsiasi lesione o stress può portare il paziente alla morte.
Nell'intestino, l'intestino tenue è più spesso colpito. L'amiloide si deposita lungo lo stroma reticolare della mucosa, sotto la membrana dei piccoli vasi, che successivamente porta all'atrofia e all'ulcerazione della mucosa. C'è una violazione dell'assorbimento, l'esaurimento si sviluppa a causa della diarrea.
Con la lipidosi si verifica una violazione dello scambio di grassi neutri, colesterolo o suoi esteri. L'obesità o l'obesità è un aumento della quantità di grassi neutri nei depositi di grasso. Si esprime nell'abbondante deposizione di grasso nel tessuto sottocutaneo, omento, mesentere, mediastino, epicardio.
Il tessuto adiposo appare dove solitamente è assente. Di grande importanza clinica è l'obesità cardiaca sviluppata. Il tessuto adiposo cresce sotto l'epicardio, avvolge il cuore, cresce nello stroma miocardico e porta all'atrofia delle cellule muscolari. Potrebbe verificarsi una rottura del cuore.
L’obesità si divide in:
1) per eziologia - in primario (idiopatico) e secondario (alimentare, cerebrale, endocrino ed ereditario);
2) secondo manifestazioni esterne - sui tipi di obesità simmetrica, superiore, media e inferiore;
3) in eccesso di peso corporeo - I grado (BMI 20–29%), II grado (30–49%), III grado (50–99%), IV grado (fino al 100% o più).
La violazione del metabolismo del colesterolo e dei suoi esteri è alla base dell'aterosclerosi. Allo stesso tempo, nell'intima delle arterie, si accumulano non solo il colesterolo e i suoi esteri, ma anche lipoproteine a bassa densità e proteine del plasma sanguigno, il che è facilitato dall'aumento della permeabilità vascolare.
L'accumulo di sostanze macromolecolari porta alla distruzione dell'intima, si disintegra e saponifica. Di conseguenza, nell'intima si formano detriti grasso-proteici, il tessuto connettivo cresce e si forma una placca fibrosa che restringe il lume del vaso.
Nelle distrofie stromali-vascolari dei carboidrati, l'equilibrio delle glicoproteine e dei glicosaminoglicani è disturbato. Le fibre di collagene vengono sostituite da una massa simile al muco. Le cause sono la disfunzione delle ghiandole endocrine e l'esaurimento. Il processo può essere reversibile, ma la sua progressione porta alla colliquazione e alla necrosi del tessuto con formazione di cavità piene di muco.
Distrofie miste. Si parla di distrofie miste nei casi in cui le manifestazioni morfologiche del metabolismo alterato si accumulano sia nel parenchima che nello stroma, la parete dei vasi sanguigni e dei tessuti. Si verificano quando si verifica una violazione del metabolismo delle proteine complesse: cromoproteine, nucleoproteine e lipoproteine, nonché minerali.
1. Violazione dello scambio di cromoproteine (pigmenti endogeni). I pigmenti endogeni nel corpo svolgono un ruolo specifico:
a) l'emoglobina svolge il trasporto dell'ossigeno - funzione respiratoria;
b) la melanina protegge dai raggi UV;
c) la bilirubina è coinvolta nella digestione;
d) la lipofuscina fornisce energia alla cellula in condizioni ipossiche.
Tutti i pigmenti, a seconda della fonte di formazione, sono suddivisi in emoglobinogenici, proteogenici e lipidogenici. I pigmenti dell'emoglobina sono costituiti da ferritina, emosiderina e bilirubina.
L'emosiderina è un pigmento che si forma in una piccola quantità in condizioni normali durante il naturale invecchiamento dei globuli rossi e il loro decadimento.
I prodotti di decadimento degli eritrociti vengono catturati dalle cellule del sistema reticoloendoteliale del fegato, della milza, del midollo osseo e dei linfonodi, dove si presentano sotto forma di granuli marroni di emosiderina. Formato in sideroblasti che contengono siderosomi. La base dell'educazione è la ferritina (proteina del ferro), che si forma quando combinata con le mucoproteine della cellula. I sideroblasti possono trattenerlo, ma ad alte concentrazioni le cellule vengono distrutte e il pigmento entra nello stroma. La ferritina viene rilevata dalla reazione Perls (il sale giallo del sangue in combinazione con l'acido cloridrico diventa blu o blu-verdastro). È l'unico pigmento contenente ferro. La sintesi di questo pigmento viene effettuata in una cellula vivente e funzionante. Si parla di violazione di questo pigmento quando la sua quantità aumenta bruscamente.
Esistono emosiderosi generali e locali. L'emosiderosi generale si verifica con l'emolisi intravascolare dei globuli rossi. Cause: varie infezioni (sepsi, malaria, ecc.), intossicazione (sali di metalli pesanti, fluoro, arsenico) e malattie del sangue (anemia, leucemia, trasfusione di sangue incompatibile con il gruppo o fattore Rh). Allo stesso tempo, gli organi sono ingranditi in volume, compattati, marroni o arrugginiti in sezione.
Alla microscopia del fegato, l'emosiderina si trova nelle cellule del sistema reticoloendoteliale in fasci lungo i seni, così come negli epatociti, cioè nel parenchima. Se il processo è insignificante, è possibile un completo recupero strutturale e funzionale e, con una gravità significativa del processo, sclerosi e, come stadio finale, cirrosi. L'emosiderosi locale si sviluppa con la rottura dei globuli rossi al di fuori del letto vascolare, cioè nei focolai di emorragie. Le più importanti sono 2 localizzazioni dell'emosiderosi: nella sostanza del cervello e dei polmoni.
Esistono 2 tipi di emorragie:
1) carattere piccolo, diapedetico; il tessuto cerebrale viene preservato, non distrutto, quindi l'emosiderina si formerà sia al centro che alla periferia del focolaio emorragico; nella sostanza della microglia cerebrale e in un piccolo numero di leucociti;
2) tipo di ematoma - quando le pareti dei vasi sanguigni sono rotte e sono accompagnate dalla distruzione della sostanza cerebrale; inoltre si forma una cavità (cisti) con pareti marroni (arrugginite); con tali emorragie, l'emosiderina si forma solo alla periferia della parete della cisti.
L'emosiderina appare nel focolaio dell'emorragia solo alla fine del 2o - inizio del 3o giorno. Un'emorragia in cui non è presente si chiama fresca, mentre dove è presente si chiama vecchia. Emosiderosi dei polmoni o indurimento marrone dei polmoni, poiché nel polmone sono combinate emosiderosi e sclerosi.
Con la pletora venosa cronica nella circolazione polmonare, si verifica l'ipossia, che porta alla diapedesi delle emorragie nel tessuto polmonare. Il pigmento si trova negli alveoli e nel setto interalveolare e l'ipossia provoca un aumento della produzione di collagene. Il setto interalveolare si ispessisce e si ispessisce. Lo scambio di gas e la ventilazione dei polmoni sono disturbati.
L'ematoidina si forma il 10-12 ° giorno in focolai di emorragia molto grandi e vecchi, che sono accompagnati dalla distruzione dei tessuti. Si trova sempre al centro del focolare. Quadro morfologico: cristalli o strutture romboidali di colore giallo o rosa.
La bilirubina è contenuta sotto forma indiretta, cioè associata all'albumina, oppure non coniugata. La bilirubina viene assorbita dagli epatociti del fegato, dove viene coniugata con l'acido glucuronico, e tale bilirubina diretta entra nell'intestino. Si dice che si verifichi una violazione con un aumento della sua quantità nel siero del sangue, seguito dalla colorazione della pelle e delle mucose in giallo.
Secondo il meccanismo di sviluppo, differiscono:
1) ittero emolitico o sovraepatico, le cui cause sono infezioni, malattie del sangue, intossicazione, trasfusione di sangue incompatibile;
2) ittero parenchimale o epatico - si verifica a causa di una malattia del fegato; gli epatociti non possono catturare completamente la bilirubina indiretta e coniugarla;
3) ittero meccanico o subepatico; cause - blocco dei dotti comuni o epatici, papilla di Vater; tumore della testa del pancreas, ecc.
A causa di una violazione del deflusso della bile, si verifica la colistasia, che è accompagnata dall'espansione dei capillari nei lobuli, dall'ispessimento della bile e dalla formazione di coaguli biliari. Gli epatociti iniziano ad essere infiltrati con pigmenti biliari e distrutti, e il contenuto inizia a entrare nei vasi sanguigni. Pertanto, la bilirubina diretta entra nel sangue e si verificano intossicazione e colorazione itterica. Inoltre, gli acidi biliari entrano nel flusso sanguigno, provocando prurito e piccole emorragie puntiformi, associate ad un'elevata permeabilità vascolare. Esiti: colangite (infiammazione dei capillari e dei dotti biliari) e sclerosi, e poi cirrosi epatica.
L'emomelanina, o pigmento malarico, si trova solo nella malaria, poiché è prodotta dal plasmodio malarico. Viene introdotto negli eritrociti e quindi catturato dalle cellule del sistema reticoloendoteliale. Il pigmento ha l'aspetto di grani neri. Gli organi sono ingranditi, densi, grigio-neri o in sezione ardesia. Con un eccesso di pigmento, si verifica l'aggregazione di questi grani: stasi malarica. La conseguenza della stasi colpisce il sistema nervoso centrale, ci sono aree di ischemia, seguite da necrosi e piccole emorragie. Inoltre, c'è un'emosiderosi generale e lo sviluppo dell'ittero emolitico.
La melanina è sintetizzata dai melanociti. La sintesi richiede gli enzimi tirosina e tirosinasi. La sintesi è regolata dai sistemi autonomo, endocrino e dagli stessi raggi UV. Il sistema vegetativo (simpatico) aumenta la produzione e il parasimpatico la diminuisce. Sistema endocrino: l'ormone adrenocorticotropo stimola e la melatonina deprime. Il pigmento si trova nello strato basale dell'epidermide. Il rapporto tra melanociti e tutte le cellule dello strato basale è 1: 15. Il disturbo segue il percorso di iperproduzione e ipoproduzione.
L'ipermelanizzazione, o malattia del bronzo (morbo di Addison), è una malattia acquisita in cui si riscontra un aumento della colorazione diffusa della pelle, ipotensione, adinamia e debolezza muscolare. La malattia è causata da danni alle ghiandole surrenali (tubercolosi, amiloidosi, processi oncologici). In queste condizioni, l’ACTH viene sintetizzato intensamente.
La xeroderma pigmentaria è una malattia congenita. La pelle è secca, itterica, iperemica, iperpigmentata e squamosa. Si verifica a causa della mancanza dell'enzima endonucleasi, che è coinvolto nell'utilizzo della melanina. Le ipermelanosi locali includono voglie. Questa è una malformazione congenita della pelle, caratterizzata dal fatto che nel processo di embriogenesi si verifica uno spostamento dal tubo neuroectodermico dei melanoblasti non solo all'epidermide, ma anche al derma. A volte una voglia può trasformarsi in un tumore maligno (melanoma).
Tra l'ipomelanosi si distinguono l'albinismo, la veitiligine e il leucoderma.
L'albinismo è una patologia congenita geneticamente determinata associata all'assenza o alla produzione insufficiente dell'enzima tirotinasi. Queste persone hanno pelle e capelli bianchi, occhi rossi, termoregolazione compromessa e funzione barriera della pelle. La durata della vita è breve.
La veitiligine è un'area depigmentata di forma irregolare. Questa patologia è geneticamente determinata ed è ereditaria.
Il leucoderma è un'area arrotondata di depigmentazione della pelle che si è verificata a seguito dell'esposizione a fattori patogeni sulla pelle. Presente in pazienti con sifilide, lebbra. Con questa patologia si notano lesioni cutanee con distruzione dei corpi Fatero-Pacino (recettori). Innanzitutto, la depigmentazione appare sulla pelle del collo e ricorda una collana di Venere. La depigmentazione può avvenire dopo ustioni, sostanze sintetiche, ecc.
La lipofuscina è un pigmento che assomiglia a granuli gialli ed è localizzato all'interno o in prossimità dei mitocondri. Normalmente è contenuto negli epatociti, nei cardiociti e nelle cellule gangliari, depositando ossigeno; in condizioni di ipossia - fornisce ossigeno alla cellula. In condizioni patologiche, vale a dire nelle infezioni croniche (ad esempio la tubercolosi) e nei processi oncologici, nelle cellule del fegato, del cuore e del sistema nervoso centrale, la quantità di questo pigmento aumenta notevolmente ed è localizzata nei lisosomi. La funzione di depositare e fornire ossigeno alle cellule non viene eseguita. Il fegato e il cuore diminuiscono di dimensioni, diventano molto densi, il colore diventa grigio-marrone (marrone).
Dettagli
Distrofia- un processo patologico complesso, che si basa su una violazione del metabolismo dei tessuti, che porta a cambiamenti strutturali.
trofico- un insieme di meccanismi che determinano il metabolismo e l'organizzazione strutturale di una cellula (tessuto) necessaria per svolgere una funzione specializzata.
Cause delle distrofie:
1) disturbi dell'autoregolazione cellulare, che possono essere causati da iperfunzione, sostanze tossiche, radiazioni, carenza di enzimi, ecc.
2) la disfunzione dei sistemi di trasporto che garantiscono il metabolismo e l'integrità strutturale dei tessuti causano ipossia.
3) violazione della regolazione endocrina e nervosa
Morfogenesi delle distrofie:
1) infiltrazione
Eccessivo accumulo di materia (normale, non anormale) come risultato di un'eccessiva sintesi.
Esempio: epatosi grassa del fegato, emosiderosi del rene.
2) decomposizione (fanerosi)
Disintegrazione delle ultrastrutture cellulari e della sostanza intercellulare, con conseguente interruzione del metabolismo dei tessuti e accumulo di prodotti del metabolismo disturbato nel tessuto.
3) sintesi perversa
Sintesi di prodotti anomali. Questi includono: la sintesi della proteina amiloide anormale nella cellula, la sintesi della proteina ialina alcolica da parte dell'epatocita.
4) trasformazione
La formazione di prodotti di un tipo di scambio da prodotti iniziali comuni che vanno alla costruzione di BJU.
classificazione della distrofia.
La classificazione segue diversi principi. Assegnare distrofie:
1) per predominanza cambiamenti morfologici nelle strutture tissutali: parenchimale, misto, mesenchimale (stroma-vascolare)
2) per predominanza violazioni dell'uno o dell'altro tipo di scambio: proteine, grassi, carboidrati, minerali.
3) a seconda influenza di fattori genetici: acquisito, ereditario.
4) di localizzazione: locale, generale.
Distrofie parenchimali.
Manifestazioni di disordini metabolici in cellule funzionalmente altamente specializzate.
1) Distrofia proteica parenchimale (disproteinosi)
L'essenza di tali distrofie è quella di modificare le proprietà fisico-chimiche e morfologiche delle proteine cellulari: subiscono denaturazione e coagulazione o colliquazione, che porta all'idratazione del citoplasma. In quei casi in cui i legami delle proteine con i lipidi si rompono, si verifica la distruzione delle strutture della membrana cellulare.
La violazione del metabolismo proteico è spesso combinata con disturbi della pompa Na-K: che porta all'accumulo di ioni Na e al rigonfiamento della cellula. Questo processo patologico si chiama distrofia idropica.
Tipi:
- granulare
Reversibile, sembra un accumulo di piccoli granuli proteici nel citoplasma. Gli organi aumentano di dimensioni, diventano flaccidi e opachi.
-flebo ialino
Nel citoplasma compaiono grandi gocce proteiche di tipo ialino, che si fondono tra loro e riempiono il corpo cellulare. In alcuni casi, termina con la necrosi focale della coagulazione della cellula.
Spesso si trova nei reni, raramente nel fegato e nel miocardio.
Nei reni, nello studio, l'accumulo di gocce si riscontra nei nefrociti. L'accumulo si osserva spesso nella sindrome nefrosica, poiché questa distrofia si basa sull'insufficienza dell'apparato vacuolare-lisosomiale dell'epitelio del tubulo prossimale, nel quale le proteine vengono normalmente riassorbite. Ecco perché nelle urine compaiono proteine (proteinuria) e cilindri (cilindruria).
L'aspetto non ha alcuna caratteristica.
Nel fegato, la microscopia rivela i corpi di Malory, costituiti da fibrille e ialina alcolica. L'aspetto di tali gocce è una manifestazione di una funzione sintetica perversa dell'epatocita, che si verifica nell'epatite alcolica, nella cirrosi biliare primaria. L'aspetto del fegato è diverso.
L'esito della distrofia a goccia ialina è sfavorevole, porta alla necrosi cellulare.
- distrofia idropica
Caratterizzato dalla comparsa nella cellula di vacuoli pieni di fluido citoplasmatico. Si osserva più spesso nell'epitelio della pelle e nei tubuli renali, negli epatociti e nei miociti.
Le cellule parenchimali sono ingrandite di volume, il loro citoplasma è pieno di vacuoli contenenti un liquido limpido. Quindi la cellula si trasforma in un enorme palloncino (l'intera cellula è diventata un grande vacuolo) - necrosi colliquazionale focale. L'aspetto dei tessuti cambia poco.
Un ruolo importante nel meccanismo di sviluppo è giocato da una violazione della permeabilità della membrana, che porta all'acidificazione del citoplasma, all'attivazione degli enzimi idrolitici dei lisosomi, che rompono i legami intramolecolari con l'aggiunta di acqua.
Cause: nei reni - danno al filtro renale, che porta all'iperfiltrazione, nel fegato - epatite di varie eziologie, nell'epidermide - edema, infezione.
L'esito di tale distrofia, di regola, è sfavorevole: termina con una necrosi focale della coagulazione.
- distrofia cornea
È caratterizzata da un'eccessiva formazione di sostanza corneo nell'epitelio cheratinizzante (ipercheratosi, ittiosi) o dalla formazione di sostanza cornea dove normalmente non esiste (cheratinizzazione patologica delle mucose). Le ragioni sono varie: disturbi dello sviluppo cutaneo, infiammazioni croniche, beriberi, ecc.
Risultato: a volte, quando la causa viene eliminata, il tessuto viene ripristinato, ma nei casi avanzati si verifica la morte cellulare.
- disturbi ereditari del metabolismo degli aminoacidi
Le cosiddette malattie da accumulo, che si basano su una violazione del metabolismo intracellulare di un numero di aminoacidi a causa della carenza ereditaria di enzimi metabolizzanti.
A) cistinosi. La scienza non sa ancora quale carenza di enzima porta a questa malattia. Gli AA si accumulano nel fegato, nei reni, nella milza, negli occhi, nel midollo osseo e nella pelle.
B) tirosinosi. Si verifica con una carenza di tirosina aminotransferasi. Si accumula nel fegato, nei reni, nelle ossa.
C) oligofrenia fenilpiruvica. Si verifica con una carenza di fenilalanina-4-idrossilasi e si accumula nel sistema nervoso, nei muscoli e nel sangue.
2) Degenerazioni grasse parenchimali (lipidosi)
I disturbi del metabolismo dei lipidi citoplasmatici possono manifestarsi con un aumento del loro contenuto nelle cellule dove si trovano normalmente, con la comparsa di lipidi dove solitamente non si trovano e con la formazione di grassi di composizione chimica insolita.
-disturbi del metabolismo lipidico
Nel fegato, la degenerazione grassa si manifesta con un forte aumento del contenuto di grassi negli epatociti e un cambiamento nella loro composizione. Nelle cellule del fegato compaiono prima i granuli lipidici (obesità polverizzata), poi piccole gocce (obesità a piccole gocce), che poi si fondono in gocce grandi (a goccia grande) o in un vacuolo grasso. Il fegato è ingrossato, flaccido e di colore giallo ocra. Tra i meccanismi di degenerazione grassa del fegato vi sono l'eccessivo apporto di acidi grassi negli epatociti o la loro aumentata sintesi da parte di queste cellule, l'esposizione a sostanze tossiche che bloccano l'ossidazione degli acidi grassi e la sintesi delle lipoproteine negli epatociti, l'insufficiente apporto di aminoacidi acidi necessari per la sintesi negli epatociti. Pertanto, l'IDP si verifica a seguito di: lipoproteinemia (alcolismo, diabete mellito, obesità generale), intossicazioni epatotrope (etanolo, cloroformio), malnutrizione.
La degenerazione grassa del miocardio si verifica a causa dell'ipossia e dell'intossicazione. Il meccanismo di sviluppo è associato ad una diminuzione dell'ossidazione degli acidi grassi dovuta alla distruzione dei mitocondri sotto l'influenza dell'ipossia o di una tossina. All'esame macroscopico, la dimensione del cuore risulta ingrandita, il muscolo cardiaco è giallo argilla. Il miocardio è simile alla pelle di una tigre: striature bianco-gialle. I lipidi sono determinati sotto forma di piccole goccioline.
Le cause della degenerazione grassa sono varie. Possono essere associati alla carenza di ossigeno (pertanto si trova spesso nelle malattie CCC), infezioni e intossicazioni, beriberi e alimentazione unilaterale.
L'esito della degenerazione grassa dipende dal suo grado. Se non è accompagnato da una grave disgregazione delle strutture cellulari, è reversibile.
-fermentopatia ereditaria
Si verificano a causa della carenza ereditaria di enzimi coinvolti nel metabolismo dei lipidi.
UN) malattia Gaucher nel deficit di glucocerebrosidasi. I lipidi si accumulano nel fegato, nella milza, nel midollo osseo.
B) malattia Niemann -Pika nel deficit di sfingomielinasi. Accumulo nel fegato, nella milza, nel midollo osseo.
IN) malattia sassa con deficit di galattosidasi acida.
G) malattia Normanno -Landinga nel deficit di beta-galattosidasi.
3) Distrofie parenchimali dei carboidrati
-distrofie dei carboidrati associate ad alterato metabolismo del glicogeno
Nel diabete mellito si osserva un uso insufficiente del glucosio da parte dei tessuti, un aumento del suo contenuto nel sangue e l'escrezione nelle urine. Le riserve di glicogeno nei tessuti vengono drasticamente ridotte. La sintesi del glicogeno nel fegato è disturbata, il che porta alla sua infiltrazione di grassi e alla degenerazione grassa del fegato.
Nei reni con diabete si verificano i seguenti cambiamenti: infiltrazione di glicogeno nell'epitelio dei tubuli.
- glicogenosi ereditaria
a) Tipo 1 - Malattia di Gierke - deficit di glucosio-6-fosfatasi
b) Tipo 2 - Malattia di Pompe - deficit di alfa-1,4-glucosidasi acida
c) 3 tipi - Malattia di Forbes - deficit di amil-1,6-glucosidasi
d) 4 tipi - malattia di Anderson - deficit di amilo-(1,4-1,6)-transglucosidasi
e) Tipo 5 – Malattia di McArdle – deficit di miofosforilasi
f) Tipo 6 - Malattia di Hers - deficit di fosforilasi epatica
Nelle malattie di tipo 1,2,5,6, la struttura del glicogeno non è disturbata.
-distrofie dei carboidrati associate a disturbi del metabolismo delle glicoproteine
Nelle cellule o nella sostanza intercellulare si verifica l'accumulo di mucine e mucoidi, chiamati anche sostanze mucose o simili al muco.
Molte cellule secernenti muoiono e desquamano, i dotti escretori delle ghiandole si ostruiscono con il muco, il che porta allo sviluppo di cisti.
Le cause sono varie, ma molto spesso - infiammazione delle mucose a seguito dell'azione di vari stimoli patogeni.
A volte nella pratica clinica esiste un fenomeno come la distrofia parenchimale. L'anatomia patologica li riferisce a disordini metabolici nelle cellule. In termini semplici, il processo di nutrizione e accumulo di nutrienti viene interrotto nel corpo, il che porta a cambiamenti morfologici (visivi). È possibile identificare tale patologia nella sezione o dopo una serie di test altamente specifici. Le distrofie parenchimali e stroma-vascolari sono alla base di molte malattie letali.
Definizione
Le distrofie parenchimali sono processi patologici che portano a cambiamenti nella struttura delle cellule degli organi. Tra i meccanismi di sviluppo della malattia si distinguono disturbi di autoregolazione delle cellule con carenza di energia, fermentopatia, disturbi discircolatori (sangue, linfa, interstizio, liquido intercellulare), distrofie endocrine e cerebrali.
Esistono diversi meccanismi di distrofia:
Infiltrazione, cioè trasporto eccessivo di prodotti metabolici dal sangue nella cellula o nello spazio intercellulare, a causa di un malfunzionamento dei sistemi enzimatici dell'organismo;
La decomposizione, o fanerosi, è la rottura delle strutture intracellulari, che porta a disordini metabolici e all'accumulo di prodotti metabolici sottoossidati;
Sintesi perversa di sostanze che normalmente la cellula non riproduce;
La trasformazione dei nutrienti che entrano nella cellula per costruire un tipo di prodotto finale (proteine, grassi o carboidrati).
Classificazione
I patologi distinguono i seguenti tipi di distrofie parenchimali:
1. A seconda dei cambiamenti morfologici:
Puramente parenchimale;
Stromale-vascolare;
Misto.
2. Per tipo di sostanze accumulate:
Proteine o disproteinosi;
Grassi o lipidosi;
carboidrato;
Minerale.
3. Per prevalenza del processo:
Sistemico;
Locale.
4. Al momento della comparsa:
Acquisita;
Congenito.
L'anatomia patologica determina alcune distrofie parenchimali non solo dall'agente dannoso, ma anche dalle specificità delle cellule colpite. La transizione da una distrofia all'altra è teoricamente possibile, ma praticamente è possibile solo una patologia combinata. Le distrofie parenchimali sono l'essenza del processo che si verifica nella cellula, ma solo una parte della sindrome clinica, che copre l'insufficienza morfologica e funzionale di un particolare organo.
Disproteinosi
Il corpo umano è costituito principalmente da proteine e acqua. Le molecole proteiche sono un componente delle pareti cellulari, delle membrane mitocondriali e di altri organelli, inoltre si trovano allo stato libero nel citoplasma. Di norma, questi sono enzimi.
La disproteinosi è altrimenti chiamata una patologia come la distrofia proteica parenchimale. E la sua essenza sta nel fatto che le proteine cellulari cambiano le loro proprietà, oltre a subire cambiamenti strutturali, come la denaturazione o il colliquamento. Le distrofie del parenchima proteico comprendono le distrofie a goccia ialina, idropica, cornea e granulare. I primi tre saranno scritti in modo più dettagliato, ma l'ultimo, granulare, è caratterizzato dal fatto che i granelli proteici si accumulano nelle cellule, a causa dei quali le cellule si allungano e l'organo aumenta, diventa allentato, opaco. Ecco perché la distrofia granulare è anche chiamata gonfiore opaco. Ma gli scienziati dubitano che si tratti di distrofia parenchimale. La patogenesi di questo processo è tale che le strutture cellulari ingrandite compensative possono essere considerate come granuli come risposta allo stress funzionale.
Distrofia delle gocce ialine

malattia di McArdle;
La sua malattia;
malattia di Forbes-Corey;
La malattia di Andersen.
La loro diagnosi differenziale è possibile dopo una biopsia epatica e l'uso dell'analisi istoenzimatica.
Interruzione del metabolismo delle glicoproteine

Si tratta di distrofie parenchimali causate dall'accumulo di mucine o mucoidi nei tessuti. Altrimenti queste distrofie vengono anche dette mucose o mucosiformi, per la caratteristica consistenza delle inclusioni. A volte si accumulano su vere e proprie mucine, ma solo su sostanze ad esse simili, che possono essere compattate. In questo caso stiamo parlando di distrofia colloidale.
La microscopia tissutale consente di determinare non solo la presenza di muco, ma anche le sue proprietà. A causa del fatto che i resti delle cellule, così come un segreto viscoso, impediscono il normale deflusso del fluido dalle ghiandole, si formano cisti e il loro contenuto tende a infiammarsi.
Le cause di questo tipo di distrofia possono essere molto diverse, ma molto spesso si tratta di un'infiammazione catarrale delle mucose. Inoltre, se una malattia ereditaria, il cui quadro patogenetico si adatta bene alla definizione di degenerazione mucoide. Questa è la fibrosi cistica. Sono colpiti il pancreas, il tubo intestinale, il tratto urinario, i dotti biliari, il sudore e le ghiandole salivari.
La risoluzione di questo tipo di malattia dipende dalla quantità di muco e dalla durata del suo rilascio. Meno tempo è passato dall'inizio del processo patologico, più è probabile che la mucosa si riprenda completamente. Ma in alcuni casi si verifica desquamazione dell'epitelio, sclerosi e disfunzione dell'organo interessato.
L'anatomia patologica è una scienza che studia la patomorfologia delle malattie a diversi livelli morfologici: macroscopico, anatomico, microscopico, microscopico elettronico e altri livelli dell'organizzazione strutturale del corpo.
La patoanatomia comprende due sezioni:
1. patologia generale;
2. patologia privata.
In patologia generale, vengono studiati i processi patologici generali.
1. danno;
2. circolazione;
3. infiammazione;
4. processi compensatori-adattativi;
5. tumori.
Il danno o l'alterazione è un processo patologico generale universale. Senza danno non c’è malattia.
I danni colpiscono tutti i livelli di organizzazione strutturale.
Si tratta di 8 livelli:
1. molecolare;
2. ultrastrutturale;
3. cellulare;
4. intercellulare;
5. tessuto;
6. organo;
7. sistema;
8. organismico.
Quando la struttura viene danneggiata a diversi livelli, si verifica una diminuzione della sua attività vitale.
Quando si studia lo sviluppo di malattie dovute a danni alle strutture, si distinguono due sezioni di patologia.
1. Eziologia.
2. Patogenesi.
L’eziologia è lo studio delle cause di lesioni e malattie.
La patogenesi è lo studio dei meccanismi di danno e sviluppo della malattia.
Tutti i fattori eziologici possono essere combinati in 7 gruppi:
1. Fattori fisici: alte e basse temperature termiche, meccaniche, radiazioni, vibrazioni elettromagnetiche.
2. Chimico: acidi, alcali, sostanze tossiche, sali di metalli pesanti e altri.
3. Tossine: veleni endogeni ed esogeni.
4. Infezioni.
5. Circolazione.
6. Neurotrofico.
7. Metabolico: disturbi metabolici durante la fame, beriberi, squilibrio nutrizionale.
Patogenesi
Questa sezione esamina meccanismi di danno come la natura dell'azione del fattore dannoso, che può essere:
diretto e indiretto.
Diretta è la distruzione diretta della struttura. Indiretto: distruzione attraverso fattori umorali, nervosi, endocrini e immunitari.
Viene studiata anche la profondità e la gravità del danno, a seconda della forza del fattore dannoso e della reattività delle strutture corporee.
Caratteristica del danno
Può essere reversibile e irreversibile. Nello sviluppo del danno passano diverse fasi, quando il danno dalle forme lievi passa a medio-grave, grave e, infine, alla morte della struttura. La morte della struttura denota il termine necrosi.
Un tipo di danno è la distrofia. Questa è una variante di danno quando la struttura è parzialmente distrutta, ma ancora conservata e funzionante.
Distrofia
Decifrare il termine: dis - disordine, trofismo nutrizionale. Cioè, una traduzione diretta significa un disturbo alimentare.
Una definizione dettagliata del termine distrofia.
La distrofia è un danno alle strutture cellulari e tissutali in risposta a una violazione del loro trofismo.
Il trofico è un insieme di meccanismi che assicurano l'organizzazione funzionale e strutturale delle cellule e dei tessuti in generale.
Esistono due tipi di meccanismi trofici:
1. cellulare;
2. extracellulare.
I meccanismi cellulari includono componenti strutturali dell'organizzazione cellulare che forniscono il metabolismo intracellulare. Allo stesso tempo, la cellula si presenta come un sistema autoregolante, in cui sono coinvolti gli organelli del citoplasma, dell'ialoplasma e del nucleo.
Vengono presentati i meccanismi extracellulari
1. sistemi di trasporto vasi sanguigni e linfatici;
2. sistema endocrino;
3. sistema nervoso.
Le distrofie possono essere il risultato di una violazione dei meccanismi del trofismo sia cellulari che non cellulari.
Pertanto, possiamo parlare di 3 gruppi di distrofie, a seconda dell'interruzione dell'attività dei meccanismi tofici:
1. distrofia dovuta a una violazione dei meccanismi cellulari del trofismo;
2. distrofia dovuta all'interruzione dei sistemi di trasporto;
3. distrofia dovuta alla rottura del sistema nervoso ed endocrino.
Nel primo gruppo di distrofie, il principale collegamento patogenetico è la fermentopatia.
Può essere l'assoluta assenza di enzimi, i relativi pochi enzimi.
Con la fermentopatia si sviluppano i processi di accumulo dei metaboliti precedenti e il blocco delle successive reazioni biochimiche.
L'accumulo di metaboliti è definito con il termine thesaurismosi - malattie da accumulo. Dalla parola greca thesauros - stock.
Il secondo gruppo di distrofie è associato a un'interruzione dell'attività dei sistemi di trasporto che garantiscono l'approvvigionamento di cibo e la rimozione di metaboliti dannosi.
Il principale collegamento patogenetico in questo caso è l'ipossia, una diminuzione della quantità di ossigeno.
Nel terzo gruppo di distrofie si osserva una violazione dell'attività del sistema nervoso ed endocrino. Il principale collegamento patogenetico in questo caso è la mancanza di sostanze biologicamente attive - bioattivatori - vari ormoni e mediatori.
Nello sviluppo delle distrofie si notano i seguenti processi morfogenetici e biochimici:
1. infiltrazione - accumulo di proteine, grassi, carboidrati nelle cellule e all'esterno delle cellule;
2. sintesi perversa - sintesi di sostanze insolite;
3. trasformazione - la transizione di alcune sostanze in altre - proteine in grassi, carboidrati in grassi e così via;
4. decomposizione (fanerosi) - la scomposizione dei complessi proteina-polisaccaride, complessi proteina-lipoproteina.
Classificazione delle distrofie
La classificazione si basa su 4 principi:
1. morfologico;
2. biochimico;
3. genetico;
4. quantitativo.
Secondo il principio morfologico, si distinguono tre tipi di distrofie, a seconda di ciò che è interessato principalmente: il parenchima cellulare o il mesenchima, le strutture intercellulari - i vasi dello stroma.
1. Parenchimale: sono colpite principalmente le cellule.
2. Mesenchima: le strutture intercellulari sono principalmente colpite.
3. Misto: danno simultaneo sia al parenchima che al mesenchima.
Secondo il principio biochimico, le distrofie si distinguono per una violazione del metabolismo di proteine, grassi, carboidrati, minerali, pigmenti e nucleoproteine.
Secondo il principio genetico si distinguono le distrofie acquisite ed ereditarie.
Secondo il principio quantitativo si distinguono le distrofie locali e quelle diffuse.
Il principio fondamentale è morfologico. Nell'ambito della classificazione morfologica funzionano anche altre classificazioni.
Di conseguenza, possiamo parlare di 3 tipi di distrofie:
1. Distrofia parenchimale.
2. Distrofia mesenchimale.
3. Distrofia mista.
Distrofie parenchimali
Secondo il principio biochimico si dividono in:
1. disproteinosi proteiche;
2. lipidosi grasse;
3. carboidrati.
Disproteinosi
La base di queste distrofie è una violazione del metabolismo proteico.
Esistono 4 tipi di distrofie proteiche
1. Granuloso.
2. Idropico.
3. Gocciolamento ialino.
4. Corneo.
Distrofia granulare
Sinonimi: gonfiore opaco e torbido.
Il termine granulare riflette il quadro istologico della patologia. In questo tipo di distrofie il citoplasma diventa granulare anziché omogeneo.
I termini gonfiore opaco e opaco riflettono l'aspetto dell'organo danneggiato.
L'essenza della patologia è che sotto l'influenza dell'azione di un fattore dannoso si verifica un aumento dei mitocondri, che conferiscono al citoplasma un aspetto granulare.
Nello sviluppo della distrofia si distinguono due fasi:
Compensazione;
Decompensazione.
Nella fase di compensazione, i mitocondri vengono ingranditi ma non danneggiati.
Nella fase di scompenso, i mitocondri sono ingranditi e leggermente danneggiati.
Tuttavia, il danno ai mitocondri è lieve. Quando il fattore dannoso cessa, ripristinano completamente la loro struttura.
Microscopicamente notato nel citoplasma di cellule di diversi organi, epatociti, epitelio dei tubuli renali, miocardiociti, granularità del citoplasma. Lo stato dei mitocondri è rivelato solo da studi al microscopio elettronico.
Vista macroscopica degli organi:
Il rene è un po' ingrossato, opaco, opaco al taglio.
Il fegato è flaccido, i bordi del fegato sono arrotondati.
Il cuore è flaccido, il miocardio è opaco, torbido, del colore della carne bollita.
Cause della distrofia granulare:
1. alterato afflusso di sangue agli organi;
2. infezioni;
3. intossicazione;
4. fattori fisici e chimici;
5. violazione del trofismo nervoso.
Significato e risultato: il processo è reversibile, ma con la continuazione del fattore dannoso, la distrofia granulare si trasforma in una forma più grave di distrofia.
Il significato clinico è determinato dalla scala della distrofia e dalla localizzazione. Con un danno miocardico totale, può verificarsi insufficienza cardiaca.
distrofia idropica
O acquoso. È caratterizzato dalla comparsa di vacuoli liquidi nel citoplasma.
Localizzazione: epitelio cutaneo, epatociti, epitelio tubulare renale, miocardiociti, cellule nervose, cellule della corteccia surrenale e cellule di altri organi.
Macroscopicamente il quadro non è specifico.
Microscopia: vengono rilevati vacuoli pieni di fluido tissutale.
Microscopia elettronica: indica che il fluido tissutale si accumula principalmente nei mitocondri, la cui struttura è completamente distrutta e da essi rimangono vescicole piene di fluido tissutale.
Nei casi di distrofia idropica grave, al posto della cellula rimane un grande vacuolo, pieno di fluido citoplasmatico. In questa variante della distrofia, tutti gli organelli del citoplasma della cellula vengono distrutti e il nucleo viene spinto verso la periferia. Questa variante della distrofia idropica è chiamata distrofia del palloncino.
L'esito della distrofia idropica, in particolare della distrofia con palloncino, è sfavorevole. La cellula alla fine potrebbe morire. E la funzione dell'organo danneggiato è significativamente ridotta.
Le cause della distrofia idropica sono infezioni, intossicazioni, ipoproteinemia durante il digiuno e altri fattori eziologici del danno.
Distrofia delle gocce ialine
L'essenza del processo è la comparsa di grumi proteici nel citoplasma delle cellule a seguito della distruzione degli organelli.
Localizzazione del rene, del fegato e di altri organi.
Cause: infezioni virali, intossicazione da alcol, uso a lungo termine di estrogeni e progesterone per prevenire la gravidanza.
Importanza: la funzione delle cellule e dell'intero organo è drasticamente ridotta. La cellula danneggiata poi muore.
Distrofia cornea
Si esprime nella comparsa eccessiva di sostanza corneo nell'epidermide cheratinizzante o in luoghi dove i processi di cheratinizzazione sono normalmente assenti.
Il processo può essere locale o generale.
1. malformazioni della pelle ittiosi - squame di pesce - una patologia congenita in cui si nota la cheratinizzazione dell'epidermide su una superficie significativa della pelle;
2. infiammazione cronica;
3. beri-beri;
4. infezione virale.
L'esito è spesso irreversibile per la cellula colpita: muore. Ma in generale, la malattia può essere curata se il fattore causale si ferma.
Significato: i focolai locali di aumentata cheratinizzazione non hanno un significato clinico particolare. Ma a volte dalle lesioni sulla mucosa può verificarsi la leucoplachia - macchie bianche - il cancro.
Una variante congenita comune della distrofia cornea, l'ittiosi, è incompatibile con la vita. I pazienti muoiono rapidamente.
Anche le malattie da accumulo in violazione del metabolismo degli aminoacidi appartengono alle distrofie del parenchima proteico.
I 3 tipi più comuni di patologia:
1. Fenilchetonuria.
2. Omocistinuria.
3. Tirosinosi.
Fenilchetonuria
Fenilchetonuria: la malattia è associata a una carenza dell'enzima - fenil-alanina - 4 idrolasi. Allo stesso tempo si nota l'accumulo di acido fenil-piruvico.
Clinica: demenza, convulsioni, difetti di pigmentazione capelli biondi, occhi azzurri, dermatiti, eczema, odore di topo. Sono presenti anche convulsioni epilettiformi, irritabilità, aggressività, oscuramento delle urine.
Patomorfologia:
1. Demielinizzazione della fibroglia del sistema nervoso centrale.
2. Degenerazione grassa del fegato.
3. Angiomatosi.
4. Ipoplasia del timo.
5. Scomparsa delle cellule nervose cerebrali.
6. Patologia vascolare degli occhi.
Omocistinuria (cistinosi)
1. ritardo mentale;
2. sublussazione del cristallino;
3. tromboembolia;
4. convulsioni.
Patomorfologia: distrofia e necrosi delle cellule del cervello, fegato, reni, displasia del tessuto osseo.
Tirosinosi
La malattia si basa su una carenza di tirosina transaminasi. Sono colpiti il sistema nervoso centrale, il fegato, i reni e le ossa. Spesso associato alla cistinosi. Patologia rara.
Lipidosi
I lipidi sono uno dei componenti dei complessi proteina-lipidi che costituiscono la base delle membrane cellulari.
Tipi di lipidi:
1. Fosfatidi - sono presenti ovunque, soprattutto nel sistema nervoso centrale.
2. Steridi: esteri di acidi grassi + alcoli ciclici (steroli). Classe diffusa di sostanze che svolgono un ruolo importante nel corpo (colesterolo, colesteroli).
3. Sfingolipidi: sfingomieline, cerebrosidi, gangliosidi. Sono particolarmente numerosi nel sistema nervoso centrale.
4. Cera: una classe di sostanze vicine ai grassi.
I grassi neutri si notano anche nel citoplasma, il cui deposito principale è il tessuto adiposo. Sono composti di glicerolo (alcali) e acidi grassi (acidi). I grassi istochimicamente neutri vengono rilevati su sezioni congelate utilizzando la colorazione Sudan 3. Sono colorati in rosso brillante.
Degenerazione grassa parenchimale
È localizzato nello stesso posto della distrofia proteica. Entrambe le distrofie sono spesso combinate.
L'aspetto macroscopico degli organi colpiti ha le sue caratteristiche.
Il cuore è ingrandito, i ventricoli sono dilatati (dilatazione), il miocardio è flaccido, simile all'argilla. Sotto l'endocardio sono visibili strisce gialle. Questo dipinto si chiama Tiger Heart.
Il fegato è ingrossato, di consistenza pastosa, di colore giallo ocra, al taglio sulla lama del coltello si formano accumuli sotto forma di placca di grasso.
I reni sono ingrossati, flaccidi, si notano piccole macchie giallastre sotto la capsula e sul taglio.
Immagine microscopica: nel citoplasma dei cardiomiociti vengono determinati l'epitelio dei tubuli renali, gli epatociti, le inclusioni di grasso sotto forma di gocce piccole, medie e grandi. La loro composizione biochimica è complessa. Questi possono essere grassi neutri, acidi grassi, fosfolipidi, colesterolo.
Cause della lipidosi parenchimale:
1. ipossia tissutale (soprattutto spesso nel miocardio);
2. infezioni - tubercolosi, processi suppurativi, sepsi, virus, alcol;
3. intossicazione - fosforo, arsenico, sali di metalli pesanti, alcool;
4. beri-beri;
5. fame - distrofia alimentare.
Opzioni iniziali:
1. con un processo leggermente pronunciato - la patologia è reversibile;
2. in caso di processo molto pronunciato, può verificarsi la morte cellulare - necrosi.
Significato: una diminuzione della funzione dell'organo fino allo sviluppo di insufficienza, il danno miocardico è particolarmente pericoloso e transitorio. Sviluppa insufficienza cardiaca e morte del paziente.
Lipidosi ereditaria
Il tipo più comune di malattia da accumulo.
Tipi di patologia:
1. Gangliosidosi.
2. Sfingomielinosi.
3. Glucocerebrosidosi.
4. Leucodistrofia.
1. Gangliosidosi - esistono 7 tipi di gangliosidosi a seconda delle varianti della fermentopatia. La malattia può manifestarsi durante l'infanzia e l'adolescenza. La malattia è particolarmente grave nella prima infanzia. Si chiamava l'idiozia amavrotica di Tay-Sachs. I sintomi della malattia sono cecità (amaurosi), distrofia e morte delle cellule nervose cerebrali con sviluppo di demenza (idiozia). La morte dei bambini arriva tra 2 4 anni.
2. Sfingomielinosi - carenza dell'enzima sfingomielinasi con accumulo di sfingomieline nelle cellule del cervello, fegato, milza e linfonodi. La patomorfologia della malattia è caratterizzata dalla comparsa di cellule schiumose - cellule nel cui citoplasma si accumulano sfingomieline che, una volta trasformate in alcoli ed eteri, si dissolvono durante la preparazione delle sezioni istologiche. E al loro posto nel citoplasma sono rimasti dei vuoti, che determinano l'aspetto schiumoso del citoplasma di queste cellule.
Sintomi clinici nella versione classica della malattia (malattia di Niemann-Pick): esordio - 5-6 mesi di vita, demenza, perdita di peso, ingrossamento del fegato e della milza, attacchi d'asma, crisi ipertermiche (febbre).
3. Glucocerebrosidosi (morbo di Gaucher).
La cosa principale è la carenza di glucocerebrosidasi e l'accumulo di glucocerebrosidi nel citoplasma delle cellule di vari organi.
Patoanatomia: distrofia del fegato, ingrossamento della milza, distrofia diffusa e morte delle cellule nervose della corteccia cerebrale. Sindrome emorragica - emorragie in diversi organi.
1. decorso cronico;
2. epatosplenomegalia;
3. iperpigmentazione;
4. demenza.
Opzioni di malattia:
1. viscerale cronico: inizia nell'infanzia e termina con la morte del paziente all'età di 20-50 anni;
2. prima infanzia acuta, di tipo neuroviscerale: la morte avviene all'età di 2 anni;
3. subacuto giovanile - inizia nell'adolescenza (18-20 anni) e termina con la morte del paziente in pochi anni.
4. Leucodistrofia.
Un gruppo di malattie in cui si verifica la distruzione della sostanza bianca del cervello e del midollo spinale (leuco - bianco; distrofia - distruzione, danno).
Questa è una patologia ereditaria, geneticamente determinata.
Clinica: violazione dell'attività del cervello e del midollo spinale, inclusa demenza, paralisi, ridotta attività del cuore.
Distrofie parenchimali dei carboidrati
I carboidrati sono una classe speciale di composti biochimici.
Nei tessuti viventi si distinguono i seguenti tipi di carboidrati complessi (polisaccaridi):
1. Glicogeno.
2. Mucopolisaccaridi.
3. Glucoproteine.
Pertanto, si distinguono i seguenti tipi di distrofie parenchimali dei carboidrati:
1. Glicogenosi.
2. Mucopolisaccaridosi.
3. Glucoproteinosi.
Glicogenosi
Possono essere ereditari e acquisiti.
Acquisito particolarmente spesso si verifica nel diabete mellito, quando si osserva una diminuzione del glicogeno negli epatociti, a causa della sua maggiore degradazione e conversione in glucosio, che si accumula nel sangue, nella linfa e nel fluido tissutale. C'è anche un aumento della glicosuria (il rilascio di glucosio nelle urine).
Così come l'accumulo di glicogeno nell'epitelio dei tubuli renali a causa della maggiore infiltrazione di glucosio nell'epitelio dei tubuli renali.
Glicogenosi ereditarie
Questo è un gruppo di malattie in cui non vi è una completa degradazione del glicogeno a causa di una carenza enzimatica. Il glicogeno si accumula nel citoplasma degli epatociti, dei miocardiociti, nell'epitelio dei tubuli renali, nei muscoli scheletrici e nelle cellule del tessuto ematopoietico.
Varianti cliniche e patomorfologiche della malattia:
1. Parenchimale: sono colpiti il fegato e i reni.
2. Muscolo-cardiaco: sono colpiti i muscoli scheletrici e il cuore.
3. Parenchimale-muscolare-cardiaco: colpisce il fegato, i reni, i muscoli scheletrici, il miocardio.
4. Parenchimale-ematopoietico: colpisce il fegato, i reni, la milza, i linfonodi.
Patomorfologia: gli organi sono ingranditi di dimensioni, soprattutto fegato, milza, il colore degli organi è pallido. Microscopicamente si verifica un aumento delle dimensioni delle cellule e un accumulo di glicogeno.
Caratteristiche biochimiche: il glicogeno ordinario, il glicogeno lungo e il glicogeno corto possono accumularsi nelle cellule.
Mucopolisaccaridosi
Descrizione dettagliata nella sezione distrofie mesenchimali.
Glucoproteinosi
1. Acquistato.
2. Ereditario.
1. Acquistato.
Degenerazione mucosa
Distrofia colloidale
La degenerazione della mucosa è l'accumulo di masse mucose nel citoplasma delle cellule. Si osserva nelle infezioni respiratorie, nell'asma bronchiale nell'epitelio dei bronchi, nelle cellule tumorali nel cancro della mucosa gastrica. Macroscopicamente - segni di muco, microscopicamente - l'aspetto di cellule a forma di anello (cellule il cui citoplasma è pieno di muco e il nucleo viene spinto verso la periferia e appiattito, motivo per cui la cellula assomiglia ad un anello).
La distrofia colloidale si manifesta con gozzo colloidale e cancro colloidale. L'esito del processo è lo sviluppo inverso o la morte della cellula, seguito da sclerosi e atrofia.
2. Ereditario.
Una malattia speciale è la fibrosi cistica.
Il muco è melma, il visco è colla per uccelli.
La cosa principale: l'accumulo di muco viscoso denso, prodotto dall'epitelio delle mucose del tratto respiratorio e gastrointestinale. Il risultato è la formazione di cisti e lo sviluppo di processi infiammatori e necrosi.
Lezione 3. Distrofie
1. Definizione, eziologia, classificazione, caratteristiche generali
Sotto distrofia (degenerazione, rinascita) comprendere i cambiamenti patologici negli organi derivanti da disordini metabolici in essi. Si tratta di cambiamenti qualitativi nella composizione chimica, nelle proprietà fisico-chimiche e nella morfologia delle cellule e dei tessuti del corpo associati a disturbi metabolici.
Le distrofie sono attribuite a danni o processi alternativi: si tratta di un cambiamento nella struttura delle cellule, della sostanza intercellulare, dei tessuti e degli organi, che è accompagnato da una violazione della loro attività vitale. Questi cambiamenti, come il tipo filogeneticamente più antico di processi reattivi, si verificano nelle prime fasi dello sviluppo di un organismo vivente.
I danni possono essere causati da diversi motivi. Agiscono sulle strutture cellulari e tissutali direttamente o attraverso influenze umorali e riflesse. La natura e l'entità del danno dipendono dalla forza e dalla natura del fattore patogeno, dalla struttura e dalla funzione dell'organo, nonché dalla reattività dell'organismo. In alcuni casi si verificano cambiamenti superficiali e reversibili a livello delle ultrastrutture, mentre in altri si verificano cambiamenti profondi e irreversibili, che possono provocare la morte non solo di cellule e tessuti, ma anche dell'intero organo.
La base della distrofia è una violazione del metabolismo delle cellule e dei tessuti, che porta a cambiamenti strutturali.
La causa immediata dello sviluppo delle distrofie può essere la violazione dei meccanismi sia cellulari che extracellulari che forniscono il trofismo:
1) un disturbo dell'autoregolazione cellulare (tossina, radiazioni, mancanza di enzimi) porta alla carenza di energia e all'interruzione dei processi enzimatici nella cellula;
2) l'interruzione dei sistemi di trasporto che forniscono il metabolismo e la struttura cellulare provoca l'ipossia, che è la causa principale nella patogenesi della distrofia;
3) un disturbo della regolazione endocrina del trofismo o una violazione della regolazione nervosa del trofismo porta alla distrofia endocrina o nervosa.
Esistono anche distrofie intrauterine.
Nelle distrofie, all'interno o all'esterno delle cellule, si accumulano prodotti metabolici (proteine, grassi, carboidrati, minerali, acqua), caratterizzati da cambiamenti quantitativi o qualitativi.
Tra i meccanismi morfologici che portano allo sviluppo dei cambiamenti caratteristici delle distrofie ci sono l'infiltrazione, la decomposizione, la sintesi perversa e la trasformazione.
I primi due sono i principali meccanismi morfologici della distrofia.
La morfologia caratteristica delle distrofie viene rilevata, di regola, a livello cellulare e tissutale.
I processi distrofici si osservano sia nel citoplasma e nel nucleo, sia nella sostanza intercellulare e sono accompagnati da una violazione della struttura delle cellule e dei tessuti, nonché da un disturbo della loro funzione.
La distrofia è un processo reversibile, ma può portare a cambiamenti irreversibili nelle cellule e nei tessuti, provocandone il decadimento e la morte.
In termini morfologici, le distrofie si manifestano con una violazione della struttura, principalmente dell'ultrastruttura delle cellule e dei tessuti, quando la rigenerazione è disturbata a livello molecolare e ultrastrutturale. In molte distrofie si trovano nelle cellule e nei tessuti inclusioni di “grani”, pietre o cristalli di varia natura chimica, che in condizioni normali non si verificano o il loro numero aumenta rispetto alla norma. In altri casi si osserva una diminuzione del numero dei composti fino alla loro scomparsa (grassi, glicogeno, minerali).
La struttura della cellula viene persa (tessuto muscolare - striatura trasversale, cellule ghiandolari - polarità, tessuto connettivo - struttura fibrillare, ecc.). Nei casi più gravi inizia la discomplessazione degli elementi cellulari. Il colore, la dimensione, la forma, la struttura e la struttura degli organi cambiano microscopicamente.
Il cambiamento nell'aspetto dell'organo è servito come base per chiamare questo processo rinascita o degenerazione, un termine che non riflette l'essenza dei cambiamenti distrofici.
La classificazione delle distrofie è associata al tipo di metabolismo compromesso. Esistono quindi distrofie proteiche (disproteinosi intracellulari, extracellulari e miste); grassi (mesenchimali e parenchimali), carboidrati (metabolismo del glicogeno disturbato), minerali (calcoli - calcoli, metabolismo del calcio alterato).
In base alla loro prevalenza si dividono in generali, sistemiche e locali; per localizzazione: parenchimale (cellulare), mesenchimale (extracellulare) e misto; secondo l'influenza di fattori genetici - acquisiti ed ereditari.
Le distrofie rientrano tra i processi reversibili, ma possono portare alla necrosi.
L'eziologia delle distrofie: gli effetti di molti fattori esterni ed interni (alimentazione biologicamente inadeguata, diverse condizioni di vita e sfruttamento, influenze meccaniche, fisiche, chimiche e biologiche, infezioni, intossicazioni, disturbi della circolazione sanguigna e linfatica, danni alle ghiandole endocrine e al sistema nervoso sistema nervoso, patologia genetica, ecc.).
I fattori patogeni agiscono su organi e tessuti direttamente o riflessivamente attraverso il sistema neuroumorale che regola i processi metabolici. La natura delle distrofie dipende dalla forza, dalla durata e dalla frequenza dell'esposizione a una particolare irritazione patogena sul corpo, nonché dallo stato reattivo del corpo e dal tipo di tessuto danneggiato.
Le distrofie si notano in tutte le malattie, ma in alcuni casi si verificano eternamente e determinano la natura della malattia, mentre in altri sono un processo patologico non specifico o non fisiologico che accompagna la malattia.
Il significato funzionale delle distrofie risiede nella violazione delle funzioni di base dell'organo (ad esempio, la sintesi di proteine, carboidrati, lipoproteine, con epatosi, comparsa di proteine nelle urine con nefrosi, debolezza del cuore con distrofia miocardica in pazienti affetti da afta epizootica, ecc.).
2. Distrofia proteica (disproteinosi), sua essenza e classificazione
L'essenza delle distrofie proteiche è che la proteina degli elementi tissutali nelle distrofie spesso differisce dalla norma nei segni esterni: è liquefatta o molto densa. A volte la sintesi delle proteine cambia, la loro struttura chimica viene disturbata. Spesso nei tessuti e nelle cellule si depositano prodotti del metabolismo proteico che non si trovano affatto in un corpo sano. In alcuni casi, i processi si limitano alla distruzione delle proteine che compongono la cellula, mentre in altri viene interrotta la struttura delle proteine che compongono le sostanze intercellulari. Le disproteinosi proteiche, che si verificano principalmente nelle cellule, comprendono i cosiddetti processi distrofici intracellulari: distrofia granulare, gocciolina ialina, idropica, distrofia cornea.
Le disproteinosi extracellulari comprendono la ialinosi e l'amiloidosi; a misto - una violazione del metabolismo delle nucleoproteine e delle glucoproteine.
3. Disproteinosi intracellulari, loro caratteristiche, esiti e significato per l'organismo
Distrofia granulare più comune di tutti i tipi di distrofie proteiche. Si manifesta indipendentemente o come componente del processo infiammatorio. Le cause delle distrofie granulari sono varie intossicazioni, disturbi circolatori, malattie infettive, stati febbrili, ecc. Tutti questi fattori possono ridurre i processi ossidativi e contribuire all'accumulo di prodotti acidi nelle cellule.
La distrofia granulare si verifica in molti organi, espressa più chiaramente nel parenchima: nei reni, nel muscolo cardiaco, nel fegato, quindi è anche chiamata parenchimale.
Segni patologici e anatomici: all'esame esterno l'organo è leggermente ingrandito, la forma è preservata, la consistenza è solitamente flaccida, il colore, di regola, è molto più chiaro del normale, il disegno sulla superficie tagliata è levigato.
Quando vengono tagliati, in particolare il rene, il fegato, a causa del gonfiore, i bordi di questi organi possono sporgere in modo significativo oltre i bordi della capsula del tessuto connettivo. Allo stesso tempo, la superficie tagliata è torbida, opaca, priva di lucentezza naturale. Ad esempio, il muscolo del cuore ha una somiglianza con il tipo di carne scottata con acqua bollente; questo ha dato motivo a molti ricercatori, nel descrivere i segni della distrofia granulare, di dire che il muscolo assomiglia a carne bollita. Torbidità, ottusità, gonfiore degli organi sono segni molto caratteristici di questo tipo di distrofia. Pertanto, la distrofia granulare è anche chiamata gonfiore torbido. Negli animali con una nutrizione migliorata, subito dopo l'alimentazione, a volte compaiono cambiamenti nei reni e nel fegato, gli stessi della degenerazione granulare, della torbidità, dell'ottusità, ma espressi in misura debole. Con la distrofia granulare, la cellula è gonfia, il citoplasma è pieno di granularità proteica piccola, appena percettibile. Quando un tale tessuto viene esposto ad una soluzione debole di acido acetico, la granularità (proteina) scompare e non si verifica più. Ciò indica il carattere proteico della granularità. Lo stesso si osserva nello studio delle fibre muscolari del cuore. I grani proteici compaiono nel muscolo, situati tra le fibrille. Le fibre si gonfiano e la striatura trasversale delle fibre muscolari si perde con l'ulteriore sviluppo del processo. E se il processo non si ferma qui, la fibra potrebbe disintegrarsi. Ma la distrofia granulare raramente cattura l'intero muscolo cardiaco, più spesso il processo avviene sulla superficie o all'interno del miocardio del ventricolo sinistro; ha una distribuzione focale. Le aree alterate del miocardio hanno un colore rosso-grigiastro.
In patologia, c'è un giudizio su due fasi nello sviluppo di questo processo. Alcuni credono che il gonfiore torbido sia lo stadio primario della distrofia granulare e che i fenomeni pronunciati di cambiamenti necrobiotici con necrosi cellulare siano la distrofia granulare. Una tale divisione dei processi distrofici è condizionale e non sempre giustificata. A volte, anche con gonfiore torbido dei reni, si verifica la necrosi cellulare.
L'essenza del processo nella distrofia è l'aumento della scomposizione di proteine, grassi, carboidrati con l'aspetto di un ambiente acido, con maggiore assorbimento di acqua e ritenzione di prodotti metabolici nelle cellule. Tutto ciò porta al rigonfiamento dei colloidi e al cambiamento nel tipo di un gruppo di proteine grossolane contenute nel citoplasma delle cellule di questi organi.
Cambiamenti particolarmente significativi nelle distrofie proteiche e, in particolare, nella distrofia granulare si verificano nei mitocondri. È noto che in questi organelli si verificano processi redox. Normalmente, a seconda dell'intensità dei processi redox, esiste una significativa variabilità nella forma e nelle dimensioni dei mitocondri. E in condizioni patologiche, specialmente quelle accompagnate da ipossia, i mitocondri si gonfiano, aumentano di dimensioni, le loro membrane esterne si allungano e quelle interne si allontanano l'una dall'altra e compaiono i vacuoli. In questa fase, la vacuolizzazione mitocondriale è reversibile. Con uno sviluppo più intenso e prolungato del processo, la vacuolizzazione può portare a cambiamenti necrobiotici irreversibili e necrosi.
Gli esiti della distrofia granulare dipendono dal grado di danno cellulare. Lo stadio iniziale di questa distrofia è reversibile. In futuro, se le cause che l'hanno causato non vengono eliminate, potrebbe verificarsi necrosi o un tipo più grave di disturbo metabolico: degenerazione grassa e idropica.
Con un lungo corso del processo, ad esempio con la febbre, non si verifica solo la distrofia cellulare, ma si verifica anche la necrosi. Questi ultimi sembrano aree luminose.
I cambiamenti nella distrofia granulare sono talvolta simili ai cambiamenti da cadavere. Ma con i cambiamenti cadaverici, non ci sarà gonfiore delle cellule, mentre con la distrofia granulare - gonfiore irregolare delle cellule con la presenza simultanea di aree di tessuto invariate nell'organo. Questi cambiamenti post mortem differiscono dalla distrofia granulare.
Gocciolamento ialino la distrofia è caratterizzata da una violazione del metabolismo proteico, procede nel citoplasma con la formazione di grandi gocce di carattere proteico. Inizialmente, queste gocce sono singole, piccole, il nucleo della cellula non è rotto. Con l'ulteriore azione della causa che provoca questo processo, le gocce aumentano di volume e di quantità, il nucleo si sposta, e poi, man mano che le gocce si formano ulteriormente, scompare gradualmente. I depositi proteici nel citoplasma acquisiscono un aspetto omogeneo, simile alla cartilagine ialina. I mitocondri sono gonfi o in uno stato di decadimento. Le goccioline proteiche che compaiono nelle cellule hanno una struttura ialina. I reni sono densi, lo strato corticale è grigio, opaco, le piramidi sono rossastre. Molto spesso, le cellule in questi casi acquisiscono il carattere di un gonfiore torbido, seguito dalla denaturazione delle proteine del citoplasma delle cellule. Se si verifica la morte del nucleo, ciò si riferisce alla necrosi cellulare.
La degenerazione delle goccioline ialine si osserva più spesso nell'epitelio dei tubuli renali, meno spesso nel fegato. A volte è combinato con la degenerazione grassa o con l'amiloidosi. Queste distrofie si osservano nelle malattie infettive croniche, nell'intossicazione e nell'avvelenamento del corpo.
Idropico (idropico o vacuolare) la distrofia è caratterizzata dal fatto che le cellule subiscono la dissoluzione-liquefazione. Inizialmente, i vacuoli con liquido sono visibili nel citoplasma, e talvolta nel nucleo, e con l'ulteriore sviluppo del processo, i vacuoli si fondono e l'intero citoplasma si riempie di liquido, il nucleo sembra fluttuare al suo interno, che poi si trasforma in una bolla piena di liquido. Tali cellule di solito muoiono. La sostanza fondamentale intercellulare e il tessuto connettivo si gonfiano e l'intero tessuto si liquefa. Con la distrofia dell'edema, i vacuoli sono visibili sui preparati trattati con alcol, quindi è necessario differenziare questi processi dalla colorazione per il grasso.
La distrofia idropitica si manifesta con edema, ustioni, vaiolo, afta epizootica, epatite virale, nevrosi croniche e altre malattie settiche.
L'esito della distrofia idropitica è favorevole nelle fasi iniziali e quando viene ripristinato il normale metabolismo dell'acqua e delle proteine, il processo è facilmente reversibile e le cellule acquisiscono un aspetto normale. Le cellule che si trovano in uno stato di grave idropia muoiono.
La distrofia vacuolare è determinata solo mediante esame microscopico. L'aspetto dell'organo non è cambiato, ma il colore è più chiaro del normale. La funzione degli organi, come in tutte le distrofie, è ridotta. La vacuolizzazione si verifica spesso nell'epitelio dei reni, nelle cellule del fegato, nella pelle, nei leucociti, nei muscoli del cuore e nelle cellule scheletriche e gangliari del sistema nervoso centrale.
Cheratinizzazione patologica o degenerazione cornea - formazione eccessiva (ipercheratosi) o qualitativamente compromessa (paracheratosi, ipocheratosi) di sostanza cornea.
La cheratinizzazione cellulare è un processo fisiologico che si sviluppa nell'epidermide ed è caratterizzato dalla graduale trasformazione dell'epitelio squamoso della pelle in scaglie cornee che formano lo strato corneo della pelle. La cheratinizzazione patologica si sviluppa a causa di malattie o danni alla pelle, alle mucose. Alla base di questi processi c'è la formazione eccessiva della sostanza cornea della pelle. Questo processo è chiamato ipercheratosi. A volte c'è una crescita eccessiva della sostanza corneo in luoghi insoliti - sulle mucose. A volte nei tumori, nelle cellule epiteliali, si forma una sostanza corneo in alcune forme di cancro.
La cheratinizzazione patologica differisce dalla cheratinizzazione fisiologica in quanto la cheratinizzazione dell'epitelio avviene sulla base di fattori che causano un aumento della formazione di sostanza cornea. Spesso c'è un processo di ipercheratosi di origine locale, che si verifica quando la pelle è irritata, ad esempio, un'imbracatura non adattata su un cavallo, una pressione prolungata sulla pelle provoca calli.
La paracheratosi si esprime nella perdita della capacità delle cellule epidermiche di produrre cheratoialina. Microscopicamente, in questa malattia, si rileva un ispessimento dell'epidermide a causa dell'iperplasia cellulare dello strato malpighiano e dell'eccessivo accumulo dello strato corneo. Con vapore e ipocheratosi, l'atrofia dello strato granulare è pronunciata, lo strato corneo è sciolto, con cellule scomposte che hanno nuclei a forma di bastoncello (cheratinizzazione incompleta).
Macroscopicamente, con la paracheratosi, lo strato corneo è ispessito, sciolto, con aumentata desquamazione dello strato corneo. Negli animali adulti, soprattutto nelle vacche da latte, si nota una crescita anomala del corno dello zoccolo, che perde la lucentezza e si fessura.
Con la leucoplachia, sulle mucose si formano focolai di epitelio cheratinizzato di varie dimensioni sotto forma di placche grigio-bianche in rilievo.
L'esito della degenerazione cornea dipende dal decorso della malattia di base. Quando la causa che provoca la cheratinizzazione patologica viene eliminata, il tessuto danneggiato può essere ripristinato.
4. Disproteinosi extracellulari e miste
Disproteinosi extracellulari
Ciò include processi patologici a lungo termine nella sostanza interstiziale del tessuto connettivo a causa di una violazione del metabolismo proteico.
Le cause di tali distrofie possono essere varie infezioni e intossicazioni, nonché il consumo prolungato di mangimi contenenti una quantità eccessiva di proteine.
Le disproteinosi extracellulari comprendono: gonfiore mucoide, fibrinoide, distrofia ialina (ialinosi) e amiloide (amiloidosi).
Gonfiore mucoide
Il gonfiore della mucosa è una disorganizzazione superficiale del tessuto connettivo, lo stadio iniziale dei suoi cambiamenti. Allo stesso tempo, nella sostanza principale e nelle fibre di collagene del tessuto connettivo, i complessi proteina-polisaccaridi vengono scissi e si accumulano mucopolisaccaridi acidi, che hanno le proprietà di metacromasia, colorazione bezofila e idrofilicità. Queste sostanze aumentano la permeabilità dei tessuti e dei vasi. Le fibre di collagene vengono preservate, ma la loro colorazione cambia. Se colorati con picrofucsina, non sono rossi, ma giallo-arancio. Questi cambiamenti sono accompagnati dalla comparsa di infiltrati linfocitici e istiolimphocitici, il gonfiore del muco viene rilevato solo al microscopio. Questa distrofia si verifica in vari organi, ma più spesso nelle arterie, nelle valvole cardiache, nell'endocardio e nell'epicardio. Il risultato può essere duplice: completa riparazione dei tessuti o transizione al rigonfiamento fibrinoide. Cause: varie forme di carenza di ossigeno, malattie del sistema metabolico ed endocrino.
gonfiore fibrinoide
Il gonfiore fibrinoide è caratterizzato dalla disorganizzazione del tessuto connettivo, che si basa sulla distruzione del collagene e della principale sostanza interstiziale, e su un forte aumento della permeabilità vascolare. Il processo di gonfiore fibrinoide è uno stadio più grave di disorganizzazione del tessuto connettivo rispetto al gonfiore mucoide. Il fibrinoide si osserva nello stroma dell'organo, nella parete dei vasi sanguigni. Inoltre, questo processo procede dalla disorganizzazione superficiale, cioè da cambiamenti superficiali, alla disintegrazione della sostanza collagene e della sostanza principale. All'esame istologico, le violazioni delle fibre di collagene sono molto significative. Diventano molto gonfi, la loro struttura fibrosa è disturbata, acquisiscono le proprietà della fibrina quando si colorano, quindi questo processo è chiamato fibrinoide, e vengono rilasciate anche sostanze proteiche come la fibrina. Con il rigonfiamento fibrinoide si verifica la disorganizzazione del tessuto connettivo con una ridistribuzione di proteine e mucopolisaccaridi. Inoltre, c'è una depolarizzazione dei mucopolisaccaridi, la loro dissoluzione. E a seconda della portata del processo di decadimento, compaiono varie proteine plasmatiche: albumine, globuline, fibrinogeno. Il cambiamento fibrinoide è una serie di condizioni del tessuto connettivo, che si basano sul gonfiore, sulla distruzione del collagene e sulla formazione di composti proteici patologici con mucopolisaccaridi e acido ialuronico.
Il processo fibrinoide è molto spesso irreversibile, si trasforma in sclerosi o ialinosi. Il significato del gonfiore fibrinoide sta nel fatto che le funzioni dei tessuti in cui si sviluppa questo processo sono attivate.
Ialinosi (distrofia ialina)
Con questo tipo di violazione del metabolismo proteico tra le cellule, appare una massa proteica omogenea, densa e traslucida: ialina.
Questa sostanza ha una notevole resistenza: non si scioglie in acqua, alcool, etere, acidi e alcali. Non ci sono reazioni speciali per il rilevamento della ialina. Nei preparati istologici si colora di rosso con eosina o magenta.
La ialinosi non è sempre un fenomeno patologico. Può verificarsi anche come fenomeno normale, ad esempio, nelle ovaie con involuzione del corpo luteo e atrofia dei follicoli, nelle arterie dell'utero e nel periodo postpartum, nell'arteria splenica negli animali adulti. In condizioni dolorose, la ialinosi viene solitamente osservata come il risultato di vari processi patologici. La ialinosi può essere locale e generale (sistemica).
Distrofia ialina locale
Nelle vecchie cicatrici, nelle capsule che circondano ascessi, necrosi e corpi estranei, si deposita la ialina. Lo stesso si osserva con la crescita del tessuto connettivo negli organi atrofizzati, con infiammazione interstiziale cronica, nei coaguli di sangue, nelle aderenze fibrose, nelle arterie con alterazioni sclerotiche.
Spesso la ialinosi non si manifesta durante l'esame esterno dell'organo e viene rilevata solo durante l'esame microscopico. Nei casi in cui la ialinosi è pronunciata, il tessuto diventa denso, pallido e traslucido.
La deposizione locale di ialina può avvenire nelle membrane proprie o basali di varie ghiandole (nella tiroide, nella mammaria, nel pancreas, nei reni, ecc.), Che si verifica più spesso con processi atrofici e in presenza di proliferazione del tessuto interstiziale . In questi casi, le vescicole e i tubuli ghiandolari sono circondati invece di un guscio proprio sottile, appena percettibile, da uno spesso anello omogeneo di sostanza ialina. Nelle cellule epiteliali si riscontrano fenomeni di atrofia.
La degenerazione ialina si osserva anche negli organi con rete reticolare, principalmente nei linfonodi. In questo caso, le fibre reticolari si trasformano in massicci filamenti densi, gli elementi cellulari tra loro si atrofizzano e scompaiono.
Il processo consiste nella deposizione lungo le fibre reticolari prima di un liquido, poi di una proteina addensante, che si fonde con le fibre in una massa omogenea. Nei linfonodi, questo si osserva più spesso con atrofia, infiammazione cronica e tubercolosi. Allo stesso tempo, le fibre di collagene si gonfiano, si fondono in fili omogenei. Atrofia cellulare.
Ialinosi generale
Questo processo diventa particolarmente importante quando lo acido ialino si deposita nelle pareti dei vasi sanguigni. Appare nell'intima e nel tessuto perivascolare delle piccole arterie e dei capillari. Si verifica un restringimento o una completa obliterazione del vaso a causa dell'ispessimento e dell'omogeneizzazione della parete. La media si atrofizza e viene sostituita da masse ialine.
La ialinosi dei vasi e del tessuto connettivo può verificarsi in due modi.
1. Si verifica una particolare modifica fisico-chimica della sostanza fibrosa con la sua trasformazione in una massa ialina omogenea. Le fibrille dei fasci di tessuto connettivo si gonfiano e si fondono, la fibrillarità si perde, i fasci diventano omogenei, privi di struttura. Successivamente, i fasci vicini si fondono, dando luogo alla formazione di campi ialini più estesi. Il tessuto connettivo acquisisce così una consistenza molto densa, spesso cartilaginea.
2. La ialinosi si verifica a causa dell'aumento della permeabilità dei vasi sanguigni e dei tessuti. C'è una sudorazione della proteina dal lume dei vasi, la proteina si coagula, si addensa e assume la forma di una massa densa vitrea. Questo processo è denominato impregnazione al plasma o plasmorragia.
La ialinosi, di regola, è un processo irreversibile, ad eccezione della ialinizzazione del tessuto connettivo cicatriziale, in cui è possibile l'allentamento e il riassorbimento della ialina. Se il processo è locale, non ci sono disturbi funzionali speciali. Con una ialinosi generale significativa, le funzioni degli organi, in particolare dei vasi sanguigni, vengono interrotte.
Amiloidosi (degenerazione amiloide)
Il processo consiste nella deposizione nei tessuti di una sostanza proteica, simile nella composizione chimica alle globuline (proteina amiloide). La sostanza è densa, omogenea, traslucida, resistente agli acidi, agli alcali, al succo gastrico, all'autolisi e alla putrefazione. L'amiloide è per molti versi simile alla ialina, ma differisce da essa e da altre proteine in alcune reazioni chimiche.
Reazione con iodio e acido solforico. Se la superficie tagliata di un organo che ha subito amiloidosi viene trattata con la soluzione di Lugol, le aree di accumulo di amiloide diventano rosso-marrone o marrone-marrone. Dopo la successiva esposizione all'acido solforico al 10%, l'amiloide acquisisce un colore blu-viola e dopo un po' diventa verde sporco.
· La colorazione con viola di metile e viola di genziana conferisce un colore rosso all'amiloide e viola ai tessuti.
Macchia rossa Congo. L'amiloide si colora di rosso-brunastro, mentre il tessuto è rosa pallido o non si colora affatto.
A volte queste reazioni non danno risultati positivi. Ciò è spiegato dai cambiamenti nella composizione chimica dell'amiloide. La sostanza amiloide che non colora è chiamata acroamiloide. I suoi depositi diventano simili a quelli ialini.
Con la deposizione di piccole quantità di amiloide l'aspetto dell'organo non cambia. Se il processo diventa pronunciato, l'organo aumenta, diventa denso, fragile, anemico; al taglio ha un peculiare aspetto traslucido, ceroso o untuoso. L'esame microscopico mostra che inizialmente la sostanza amiloide si deposita solitamente nelle pareti dei piccoli vasi sanguigni, sotto la membrana argirofila dell'endotelio, nonché lungo le fibre reticolari e sotto la membrana basale dell'endotelio.
La degenerazione amiloide può essere comune, diffusa, quando il processo coinvolge diversi organi. In altri casi è locale: limitato a un luogo qualsiasi.
L'amiloidosi della milza è follicolare e diffusa.
R. Nella forma follicolare, i depositi di amiloide si verificano prima lungo la periferia dei follicoli nel reticolo, per poi diffondersi all'intero follicolo. I linfociti vengono espulsi.
L'arteria centrale è ispessita, ha un aspetto omogeneo. Macroscopicamente la milza è moderatamente ingrandita. La sezione mostra follicoli alterati sotto forma di grani di sago bollito (“milza di sago”).
B. Nella forma diffusa, l'amiloide si deposita nei follicoli e nella polpa rossa. Inizialmente compaiono isole separate, di forma irregolare, che poi si fondono in una massa continua. Atrofia cellulare. La milza è ingrossata, densa (solo nei cavalli pastosi). La superficie tagliata è di colore rosso-marrone chiaro e ricorda un prosciutto ("grasso, o prosciutto, milza").
amiloidosi del fegato. I cambiamenti si diffondono dalla periferia al centro dei lobuli. Inizialmente, l'amiloide si deposita tra l'endotelio dei capillari intralobulari e i fasci epatici, nonché nelle pareti dei vasi interlobulari. Man mano che il processo aumenta, si formano aree continue di masse amiloidi e le cellule del fegato si atrofizzano. Il fegato è ingrossato, denso, marrone chiaro. Solo nei cavalli è flaccido e si strappa facilmente.
Amiloidosi dei reni. Il processo inizia con i glomeruli. L'amiloide si deposita sotto le membrane argirofile dell'intima delle arterie, delle arteriole e delle anse vascolari dei glomeruli. I grumi si accumulano, schiacciando gli anelli. A poco a poco, l'intero glomerulo viene sostituito dall'amiloide. L'amiloidosi si diffonde anche nelle pareti dei vasi della corteccia e del midollo sotto la membrana dell'epitelio tubolare. Nell'epitelio dei tubuli si verificano cambiamenti distrofici e atrofia. I reni sono ingrossati, densi, la superficie dell'incisione è cerosa. Le cause della degenerazione amiloide sono varie. Questi includono malattie infettive croniche in cui si verificano suppurazione e necrosi, come actinomicosi, tubercolosi; meno spesso si verifica nelle malattie croniche che si verificano senza suppurazione e necrosi. La causa dell'amiloidosi può essere il consumo prolungato e abbondante di cibi ricchi di proteine (ad esempio, oche da ingrasso). Di norma, questo tipo di distrofia si osserva nei cavalli - produttori di siero.
L'esito dell'amiloidosi generale è sfavorevole, poiché si verificano cambiamenti distrofici negli organi alterati, atrofia e necrosi del parenchima.
Disproteinosi miste
La disproteinosi mista è una violazione del metabolismo proteico della cellula e della sostanza intercellulare. I processi distrofici si verificano quando si verifica una violazione del metabolismo di proteine complesse: nucleoproteine, glicoproteine e cromoproteine.
Violazione del metabolismo delle nucleoproteine
Le nucleoproteine sono costituite da proteine e acidi nucleici (DNA e RNA). Il prodotto finale del metabolismo delle nucleoproteine è l'acido urico e i suoi sali. In condizioni normali, questi prodotti di decadimento allo stato disciolto vengono escreti dal corpo principalmente dai reni. In violazione del metabolismo delle nucleoproteine, si verifica un'eccessiva formazione di acido urico e i suoi sali si depositano nei tessuti; questo si osserva nella diatesi dell'acido urico e nell'infarto renale da acido urico.
La diatesi dell'acido urico è la deposizione di sali di acido urico in vari tessuti e organi. Questo si nota solitamente sulle superfici articolari delle dita delle estremità, nei tendini, nelle cartilagini del padiglione auricolare, nei reni e sui tegumenti sierosi. Nel sito di deposizione dei cristalli di sali urinari, gli elementi tissutali subiscono necrosi, attorno alle zone morte si sviluppa una reazione infiammatoria con la crescita del tessuto connettivo.
Più spesso, gli uccelli (polli, anatre) si ammalano di diatesi dell'acido urico, meno spesso - i mammiferi. Negli uccelli i sali di urato si depositano sotto forma di una densa massa biancastra sulle membrane sierose della cavità addominale, sul pericardio e sull'epicardio, nei reni e sulle superfici articolari delle dita dei piedi. Sotto le sovrapposizioni si rivela una copertura sierosa infiammata. I reni sono ingranditi, punteggiati da una patina biancastra e sulla superficie tagliata si trovano focolai grigio-biancastri o bianco-giallastri. Al microscopio sono visibili cristalli di urato radianti; l'epitelio dei tubuli renali è in uno stato di degenerazione granulare e necrosi, e lo stroma è infiltrato da cellule linfoidi e giganti. Una lesione caratterizzata dalla deposizione di sali di acido urico nelle articolazioni delle dita dei piedi è chiamata gotta. In questo caso, le articolazioni si gonfiano, si deformano e si formano nodi densi.
L'infarto renale da acido urico è una condizione fisiologica che si manifesta negli animali appena nati nei primi sette giorni, dopodiché scompare. Ciò è dovuto al cambiamento nei processi metabolici. Nel sangue aumenta temporaneamente la concentrazione di acido urico, che non ha il tempo di essere completamente escreto dal corpo con l'urina. Macroscopicamente, sulla superficie del taglio dei reni nel midollo, si trovano radialmente strisce giallo-rossastre, che sono un accumulo di sali urinari nel lume dei tubuli diretti e nello stroma dei reni. Negli animali adulti, con infiammazione e necrosi della mucosa della vescica e della pelvi renale, possono verificarsi incrostazioni (intervallate) di frammenti di acido urico nei tessuti morti.
La deposizione di acido urico negli organi provoca cambiamenti irreversibili (necrotici) nei tessuti colpiti.
Interruzione del metabolismo delle glicoproteine
Le glicoproteine sono composti complessi di proteine con polisaccaridi contenenti esosi, esosamine e acidi esuronici.
La degenerazione della mucosa come processo patologico si verifica nelle cellule epiteliali delle mucose, nelle cellule di numerose ghiandole e nel tessuto connettivo. È una conseguenza di una violazione del metabolismo delle glucoproteine ed è caratterizzata dall'accumulo di mucine e mucoidi nelle cellule. Nell'epitelio, la degenerazione della mucosa può essere il risultato dell'ipersecrezione delle ghiandole mucose con aumento della desquamazione delle cellule epiteliali e della loro trasformazione in una massa simile al muco. Nel tessuto connettivo della degenerazione mucosa è esposta una sostanza interstiziale, nella quale si accumulano sostanze mucoidi.
In presenza di acqua il muco si gonfia e quando viene aggiunto acido acetico o alcool precipita e cade sotto forma di una rete fibrosa sottile e delicata. In questo, il muco differisce dalle sostanze simili al muco (mucoidi) che si formano nei tessuti sia in condizioni normali che patologiche. Il muco, come l'amiloide, ha metacromosia. Pertanto, quando vengono colorati con il viola cresilico, la tionina, il tessuto normale si colora di blu e il muco si colora di rosso.
La degenerazione mucosa delle cellule epiteliali è ben espressa nelle infiammazioni catarrali delle mucose, in particolare degli organi respiratori e digestivi. In condizioni fisiologiche, la secrezione del muco - un prodotto della secrezione delle cellule caliciformi - avviene come segue. Innanzitutto, nelle cellule compaiono le più piccole goccioline trasparenti di muco che, fondendosi tra loro, formano gocce di dimensioni maggiori. La cellula aumenta di volume, si gonfia e alla fine il muco viene versato sotto forma di segreto, dopodiché la cellula crolla e ripristina il suo aspetto precedente. Quindi su di esso iniziano di nuovo ad apparire goccioline di muco.
La degenerazione mucosa delle cellule epiteliali è accompagnata da una maggiore formazione e separazione di muco, necrosi e rigetto delle cellule epiteliali morte, i cui resti sono mescolati con muco.
La degenerazione della mucosa può colpire vari tipi di tessuto connettivo, comprese la cartilagine e le ossa, nonché tumori del tipo del tessuto connettivo. Nel tessuto connettivo si osserva gonfiore e, per così dire, dissoluzione delle fibrille.
Nelle ossa con degenerazione mucosa, prima scompare il calcare e poi la sostanza osteoide si liquefa. Al microscopio il tessuto alterato appare come una massa omogenea e priva di struttura, dalla quale cadono filamenti di mucina sotto l'azione di acidi e alcol. La causa più comune che contribuisce alla comparsa della distrofia della mucosa del tessuto connettivo è una violazione del trofismo dei tessuti nelle malattie infettive croniche, intossicazioni, disturbi delle ghiandole endocrine e tumori.
Una volta eliminate le cause della degenerazione della mucosa, il tessuto viene ripristinato.
5. Violazione dello scambio di cromoproteine (pigmenti). Pigmenti esogeni ed endogeni
Tutti i tessuti e gli organi degli animali sono caratterizzati da un certo colore: pigmentazione. Alcuni pigmenti si trovano nei tessuti allo stato disciolto, mentre altri si presentano sotto forma di depositi granulari, amorfi e cristallini. Tutti sono formati dal corpo stesso, si trovano in condizioni fisiologiche e sono chiamati endogeni. Inoltre, in determinate condizioni patologiche, i pigmenti dell'ambiente esterno, normalmente non caratteristici di esso, possono penetrare nel corpo di un animale e di una persona. Si chiamano esogeni.
I pigmenti endogeni si suddividono a loro volta in tre gruppi a seconda della fonte della loro formazione.
1. Emoglobinogenici, che derivano dall'emoglobina durante le sue varie trasformazioni. Questi includono ferritina, ematoidina, emosiderina e bilirubina studiati in patomorfologia.
2. Pigmenti proteinogenici che non sono correlati all'emoglobina e sono derivati della tirosina e del triptofano. Questi includono la melanina, gli andrenocromi e il pigmento delle cellule enterocromaffini.
3. Pigmenti lipidogenici associati al metabolismo dei grassi. Questi includono lipocromi, lipofuscina e ceroide.
Pigmenti emoglobinogenici si formano a seguito della rottura fisiologica e patologica degli eritrociti, che includono l'emoglobina cromoproteica ad alto peso molecolare, che conferisce al sangue un colore specifico.
La ferritina è una proteina di riserva del ferro. Si forma dal ferro alimentare nella mucosa intestinale e nel pancreas e durante la degradazione dei globuli rossi e dell'emoglobina nella milza, nel fegato, nel midollo osseo e nei linfonodi. In questi organi può essere isolato mediante reazione istochimica allo smalto prussiano.
L'emosiderina è un pigmento contenente ferro amorfo a grana fine di colore marrone dorato o marrone. Si trova a livello intracellulare e in caso di decadimento cellulare giace liberamente nei tessuti. L'emosiderina è formata dalle cellule durante la fagocitosi degli eritrociti o dall'emoglobina disciolta nel plasma. La comparsa di emosiderina nei tessuti è detta emosiderosi, che può essere generale e locale.
L'emosiderosi generale si verifica con emolisi intravascolare, ad esempio con sepsi, anemia infettiva dei cavalli, piroplasmosi e con alcuni avvelenamenti (arsenico, fosforo, ecc.). L'emoglobina rilasciata dagli eritrociti si dissolve nel plasma ed è parzialmente escreta nelle urine. L'altra parte viene assorbita dalle cellule reticoloendoteliali e si trasforma in emosiderina, si verifica un'emosiderosi generale. L'emosiderina si forma solo a livello intracellulare. I depositi di emosiderina si verificano principalmente nella milza, poi nel fegato, nel midollo osseo, nei linfonodi e anche nei reni in ordine alla funzione escretoria. Questi organi assumono emosiderina, si trovano nelle cellule reticoloendoteliali e nell'epitelio dei tubuli contorti dei reni. L'emosiderina è solubile negli acidi, insolubile negli alcali, nell'alcool e nell'etere, non scolorisce con il perossido di idrogeno. Le seguenti reazioni vengono utilizzate per differenziare l'emosiderina da altre inclusioni intracellulari.
Reazione Perls: quando le sezioni istologiche vengono trattate con ferricianuro di potassio (sale giallo del sangue) in presenza di acido cloridrico, il pigmento diventa blu-verdastro (“smalto di Berlino”).
Dall'aggiunta di solfuro di ammonio, l'emosiderina diventa nera e, con un ulteriore trattamento con ferricianuro di potassio e acido cloridrico, il pigmento diventa blu ("blu turnbull").
L'emosiderosi locale si nota con emolisi extravascolare degli eritrociti, che si osserva con emorragie. L'emosiderina si accumula nel citoplasma delle cellule lungo la periferia dell'emorragia.
L'ematoidina si forma anche dalla degradazione dell'emoglobina. Questo pigmento non contiene ferro, ha la forma di cristalli che sembrano formazioni rombiche o assomigliano a fasci di aghi di colore arancione brillante. Con l'accumulo di pigmento, appaiono varie figure sotto forma di stelle, pannocchie, covoni, ecc. Meno comunemente, l'ematoidina si presenta sotto forma di grani o grumi amorfi. Questo pigmento si dissolve negli alcali, si decompone con forti acidi nitrico e solforico, è difficile da sciogliere in alcool ed etere e non scolorisce con il perossido di idrogeno.
L'ematoidina si forma nelle parti centrali delle emorragie, dove non ci sono cellule e accesso all'ossigeno.
Bilirubina. Questo pigmento si forma costantemente e subisce costantemente varie trasformazioni, partecipando al metabolismo di un organismo normale. Si forma nel sistema reticoloendoteliale durante la distruzione fisiologica degli eritrociti, entra nel fegato e lì viene incluso nella bile formata dalle cellule epatiche. La bilirubina viene disciolta nella bile e provoca la sua caratteristica colorazione. Nelle sue proprietà, questo pigmento è vicino all'ematoidina e dà una reazione positiva alla Gmelina: quando esposto all'acido nitrico, si formano anelli colorati. Normalmente, la bile si trova nei dotti biliari e nella cistifellea, da dove viene espulsa nel duodeno. In condizioni patologiche, la normale formazione e secrezione della bile è disturbata; la bilirubina entra nel flusso sanguigno, che è accompagnato dalla colorazione dei tessuti in giallo. Una tale colorazione gialla di tutti gli organi, e in particolare della sclera degli occhi, delle mucose visibili, dei tegumenti sierosi e dell'intima dei vasi sanguigni, è chiamata ittero, che è diviso in tre tipi per origine e patogenesi: emolitico, parenchimale e meccanico.
· L'ittero emolitico si verifica con l'emolisi intravascolare dei globuli rossi. Grandi quantità di prodotti di degradazione dell'emoglobina entrano nelle cellule del sistema reticoloendoteliale. Pertanto, viene prodotta intensamente la bilirubina o un pigmento ad essa vicino, che entra direttamente nel sangue.
L'ittero parenchimale è causato da una violazione del deflusso della bile dal fegato, che si verifica a causa della disfunzione delle cellule epatiche. Queste cellule perdono la capacità di secernere la bile nei capillari biliari, quindi la bile si diffonde nel sangue attraverso le pareti dei capillari sanguigni e linfatici. Le cause dell'ittero parenchimale sono varie. Si tratta principalmente di malattie infettive e avvelenamenti.
Pigmenti proteogenici includono melanina, andrenocomi e pigmento cellulare enterominico.
Melanina: questo pigmento determina il colore della pelle, dei capelli, del piumaggio degli uccelli, degli occhi. Normalmente il contenuto di melanina dipende dal tipo di animale, dalla razza, dall'età e dalle sue caratteristiche individuali. Microscopicamente, la melanina si trova sotto forma di granelli marroni o neri che giacciono nel protoplasma delle cellule. Chimicamente, la melanoproteina contiene zolfo, carbonio e azoto, ma è priva di ferro e grassi. Non si dissolve negli acidi e negli alcali, diventa nero con il nitrato d'argento e scolorisce per l'azione dell'acqua ossigenata. La formazione della melanina avviene nelle cellule dello strato malpighiano dell'epidermide e della retina. Le cellule che producono melanina sono chiamate melanoblasti.
Le violazioni della melanogenesi si manifestano con una maggiore formazione di melanina, il suo accumulo in luoghi insoliti, la scomparsa o l'assenza di pigmento. Questi disturbi possono essere acquisiti o congeniti e possono essere diffusi o locali.
L'eccessiva formazione di melanina nella pelle e il suo deposito negli organi interni è chiamata melanosi generale. Si verifica più spesso nei bovini e nei piccoli bovini, soprattutto nei vitelli e nelle pecore. Si ritiene che questo processo sia di origine foraggera. La melanina si deposita nel fegato, nei polmoni e sulla pelle sierosa, meno spesso nelle membrane del cervello e del midollo spinale, che diventano marrone scuro o marrone-nero.
L'eccessiva pigmentazione locale della pelle è associata a una crescita eccessiva benigna o maligna di melanoblasti con formazione di melanomi.
Si verificano spesso nei cavalli e nei cani grigi. Le fonti del loro aspetto sono voglie.
La formazione insufficiente congenita di melanina o la sua completa assenza nel corpo è chiamata albinismo. Questa condizione è caratteristica di alcune specie e razze di animali (topi bianchi, ratti, conigli, ecc.).
La depigmentazione congenita locale della pelle viene definita vitiligine. In alcuni casi, dopo un'infiammazione prolungata e altre lesioni (ferite, ulcere, malattie accidentali dei cavalli), si formano sulla pelle macchie non pigmentate, chiamate leucoderma.
pigmenti lipidogenici. Questi includono lipocromi, lipofuscina e ceroide. Contengono sostanze grasse e proteiche.
La lipofuscina è una glicolipoproteina che assomiglia a grani o grumi di colore marrone. La sua formazione è associata a un processo ossidativo: autossidazione di fosfolipidi e grassi. Colorato di rosso con Sudan III e scarlatto, non reagisce al ferro. Non si dissolve negli acidi e negli alcali; dal nitrato d'argento, a differenza della melanina, non annerisce. Negli animali, la lipofuscina si trova nei muscoli cardiaci, scheletrici e lisci, nei reni, nelle ghiandole surrenali, nel fegato, nelle cellule nervose, nelle vescicole seminali e nei testicoli.
La pigmentazione patologica con lipofuscina si manifesta solitamente con atrofia dei muscoli cardiaci, del fegato, dei reni e delle cellule del sistema nervoso centrale.
I pigmenti di emofuscina, presenti nel fegato di cavalli affetti da encefalomielite infettiva, e il ceroide, la cui formazione è associata all'ipovitaminosi E, sono identici nella composizione fisico-chimica alla lipofuscina.
I lipocromi sono pigmenti gialli che conferiscono un colore giallo al tessuto adiposo, alla corteccia surrenale, al tuorlo d'uovo, al siero del sangue, ecc. I lipocromi includono anche la luteina, il pigmento del corpo luteo ovarico. Questi pigmenti si dissolvono nei reagenti - solventi grassi e sono un lipide in cui si dissolvono idrocarburi colorati - carotenoidi e flavine. La formazione di lipocromo e luteina è associata al metabolismo dei grassi e delle proteine. Con l'atrofia del tessuto adiposo negli animali anziani e malnutriti, il grasso acquisisce un ricco colore giallo.
pigmenti esogeni
Questo è il nome di varie sostanze colorate che entrano nel corpo dall'ambiente esterno, che possono modificare il colore naturale degli organi o conferire loro una tonalità diversa. Molto spesso, si osservano pigmenti esogeni nei polmoni, nei linfonodi regionali, meno spesso nella milza, nel fegato e nei reni. La deposizione di materiali estranei nei polmoni è chiamata pneumoconiosi. Ciò si può osservare quando gli animali sostano per lungo tempo in luoghi dove l'aria è inquinata da particelle di polvere di varia origine. La più importante è la spolveratura dei polmoni con polvere di carbone: antracosi.
Sotto la pleura e all'interno dei lobi polmonari si trovano accumuli di carbone sotto forma di aree nere o sotto forma di spolvero diffuso. Al microscopio sono visibili particelle di carbone attorno ai vasi sanguigni, nell'epitelio alveolare e nell'interstizio. La polvere di carbone si accumula anche nei linfonodi mediastinici e bronchiali. Con una significativa deposizione di particelle di carbone, la loro azione può causare alterazioni infiammatorie nei polmoni, seguite dalla proliferazione del tessuto connettivo. Quando i polmoni vengono spolverati di particelle calcaree, compaiono focolai biancastri (calicosi). Se i polmoni diventano polverosi di silice, allumina o grumi di quarzo, si verifica la silicosi, che è accompagnata dalla sclerosi dei polmoni.
In caso di trattamento prolungato degli animali con farmaci contenenti argento, quest'ultimo si deposita nell'epitelio dei glomeruli vascolari, nella membrana basale dei tubuli renali (argirosi renale). I sali d'argento si trovano anche nel fegato, nelle cellule di Kupffer e nelle pareti dei vasi sanguigni. Macroscopicamente, i tessuti affetti da argirosi acquisiscono un colore grigio (acciaio).
Come puoi mangiare troppo prima della distrofia. Il nostro corpo è costituito da tessuti diversi, ma due di essi sono noti a tutti: grasso corporeo e muscoli. Si è parlato molto del grasso corporeo, e non tutto è dei migliori, anche dei muscoli - solo con l'atteggiamento più benevolo. Questi sono due
Dal libro Farmacologia clinica omeopatica autore Ernest FarringtonLEZIONE 33 Rubiaceae - Robbia Rubiaceae:1. Rubia titctoiria (Robbia) .2. Galium (anche vernice rossa).3. China.4. Ipecacuanha.5. Caffè.6. Mitchella.7. Gambier Oggi disponiamo di una famiglia di piante dalla quale ricaviamo tre rimedi molto preziosi, Cinchona, Ipecacuanha e Coffea. Questa famiglia ci dà anche Gambier (Gambogia,
Dal libro Storia della medicina: appunti delle lezioni autore E. V. BachiloLEZIONE 35 Scrophulariaceae - Norichaceae Cina. Da questa famiglia di piante si ottengono Digitalis, Gratiola, Leptandra viginica, Euphrasia, Verbascum e Linaria. Abbiamo pochi sintomi per ciascuno di questi rimedi e quelli conosciuti sono sufficientemente specifici da essere facili da ricordare. Il più importante
Dal libro Anatomia patologica autore Marina Aleksandrovna KolesnikovaLEZIONE 37 Solanaceae - Nightshade Solanaceae:1. Belladonna.2. Iosciamo.3. Stramonio.4. Solanum nigr.5. Tabacco.6. Dulcamara.7. Capsicum I rimedi che compongono questo gruppo sono molto simili tra loro nella loro sintomatologia. Non c'è quasi un solo sintomo di questi rimedi che non si presenti quasi nella stessa forma.
Dal libro Omeopatia pratica autore Viktor Iosifovich VarshavskijLEZIONE 42 Gruppo Minerale Nella tabella allegata ho posto per il tuo studio gli elementi nel loro rapporto reciproco, in una certa misura proprio come lo troviamo in chimica. Pertanto, non sono disposti nell'ordine accettato in farmacologia. Ma non è assoluto.
Dal libro Anatomia patologica generale: appunti di lezione per le università autore GP DemkinLEZIONE N. 1. Lezione introduttiva. Simboli medici di tempi e popoli diversi La storia della medicina è la scienza dello sviluppo, del miglioramento delle conoscenze mediche e delle attività mediche di diversi popoli del mondo nel corso della storia dell'umanità, che è
Dal libro Esercizi meditativi per ripristinare la vista secondo il metodo del professor Oleg Pankov autore Oleg Pankov3. Distrofie parenchimali e proteiche Le distrofie parenchimali si dividono in proteine, grassi e carboidrati.La distrofia proteica è una distrofia in cui il metabolismo delle proteine è disturbato. Il processo di distrofia si sviluppa all'interno della cellula. Tra le proteine del parenchima
Dal libro Allenamento e giochi per i muscoli oculari. Esercizi unici per ripristinare la vista secondo il metodo del professor Oleg Pankov autore Oleg Pankov6. Distrofie miste Si dicono distrofie miste quei casi in cui le manifestazioni morfologiche di metabolismo alterato si accumulano sia nel parenchima che nello stroma, la parete dei vasi sanguigni e dei tessuti. Si verificano quando si verifica una violazione del metabolismo delle proteine complesse: cromoproteine,
Dal libro Riprendi la vista. Lezioni sul ripristino naturale della vista autore Vladimir Georgievich ZhdanovDIFETTI DEL MUSCOLO CARDIACO (DIFETTI CARDIACI, DISTROFIA MIOCARDIACA, ATEROSCLEROSI, MANCANZA DI CIRCOLAZIONE) Arnica 3X, 3 - con ipertrofia miocardica causata dal suo sovraccarico Aurum - ipertrofia miocardica con ipertensione, aterosclerosi Barita carbonica 3, 6, 12 -
Dal libro dell'autoreConferenza 5. Distrofie minerali 1. Rachitismo, osteomalacia, osteodistrofia fibrosa 2. Calcoli e calcoli, loro caratteristiche morfologiche, composizione chimica e significato per l'organismo animale 3. Distrofie da carboidrati
Dal libro dell'autoreCorrezione della vista con miopia, astigmatismo, distrofia retinica, glaucoma. Questi esercizi contribuiscono allo sviluppo della sensibilità della fossa centrale (macula) della retina, aumentano l'acuità visiva e migliorano l'afflusso di sangue agli occhi, allenano tutti e sei i muscoli oculari e
Dal libro dell'autoreAllenamento "Segno nell'ovale" con miopia, distrofia retinica, glaucoma, strabismo Questi esercizi aiutano a sviluppare la sensibilità della fossa centrale (macula) della retina, aumentano l'acuità visiva e migliorano l'afflusso di sangue agli occhi.
Dal libro dell'autoreLezione 1 Salve di nuovo, cari colleghi, stiamo iniziando la nostra prima lezione presso l'Università Popolare su uno stile di vita sano utilizzando il metodo di Gennady Andreyevich Shichko. Il corso sarà incentrato sulla correzione della vista utilizzando il metodo Shichko-Bates. Questo è un corso generale di salute e benessere.
Dal libro dell'autoreLezione 2 Allora, ciao cari colleghi, iniziamo la nostra seconda lezione presso l'Università Popolare su uno stile di vita sano secondo il metodo di Gennady Andreyevich Shichko. Il corso è dedicato alla correzione della vista mediante il metodo Shichko-Bates. Questo è un corso di guarigione generale e di eliminazione dei danni
Dal libro dell'autoreLezione 3 Ciao, cari colleghi, stiamo iniziando la nostra terza lezione presso l'Università Popolare su uno stile di vita sano secondo il metodo di Gennady Andreyevich Shichko. Il corso è dedicato alla correzione della vista mediante il metodo Shichko-Bates. Questo è un corso di guarigione generale e di eliminazione dei danni
- In contatto con 0
- Google Plus 0
- OK 0
- Facebook 0






