L'articolo fornisce un'idea di base di come vengono creati i farmaci nel mondo moderno. Vengono presi in considerazione la storia della progettazione dei drag, i concetti di base, i termini e le tecnologie utilizzate in quest'area. Particolare attenzione è rivolta al ruolo della tecnologia informatica in questo processo ad alta intensità scientifica. Vengono descritti i metodi per la ricerca e la validazione di bersagli biologici per i farmaci, lo screening ad alto rendimento, i processi per le sperimentazioni cliniche e precliniche sui farmaci, nonché l'uso di algoritmi informatici.
Drag design: la storia
L'industria della progettazione diretta di nuovi farmaci, o, come viene chiamato questo processo, derivando dall'inglese per mancanza dello stesso termine russo breve e conveniente, progettazione di farmaci ( farmaco- medicinale, progetto- progettazione, costruzione) è una disciplina relativamente giovane, ma ancora non così giovane come comunemente si crede.
Figura 1. Paul Ehrlich, che per primo ipotizzò l'esistenza dei chemocettori e il loro possibile utilizzo in medicina.
Biblioteca nazionale di medicina degli Stati Uniti
Alla fine del XIX secolo la chimica aveva raggiunto un notevole grado di maturità. Fu aperta la tavola periodica, furono sviluppate la teoria della valenza chimica, la teoria degli acidi e delle basi e la teoria dei composti aromatici. Questo indubbio progresso ha dato slancio alla medicina. Nuovi prodotti chimici - coloranti sintetici derivati da resine - iniziarono ad essere utilizzati in medicina per la colorazione differenziale dei tessuti biologici. Nel 1872-1874 a Strasburgo, nel laboratorio del famoso anatomista Wilhelm Waldeer, lo studente di medicina Paul Ehrlich (Fig. 1), che studiò la colorazione selettiva dei tessuti, ipotizzò per primo l'esistenza di chemocettori - speciali strutture tissutali che interagiscono specificamente con le sostanze chimiche , e ha postulato la possibilità di utilizzare questo fenomeno nel trattamento di varie malattie. Successivamente, nel 1905, questo concetto fu ampliato da J. Langley, che propose un modello del recettore come generatore di impulsi biologici intracellulari, che viene attivato dagli agonisti e inattivato dagli antagonisti.
Questo momento può essere considerato la nascita della chemioterapia e una nuova pietra miliare nella farmacologia, e nel XX secolo ha portato a un successo senza precedenti nella medicina clinica. La penicillina, un antibiotico scoperto nel 1929 da Alexander Fleming e successivamente studiato da Chain e Flory, può essere giustamente definita una delle conquiste di più alto profilo dell'industria farmaceutica del 20° secolo. La penicillina, che ha un effetto antibatterico, ha reso un servizio indispensabile all'umanità durante la seconda guerra mondiale, salvando la vita a milioni di feriti.
Impressionate dal successo della penicillina, molte aziende farmaceutiche hanno aperto le proprie divisioni di microbiologia, riponendo in loro la speranza di scoprire nuovi antibiotici e altri farmaci. I successivi progressi della biochimica hanno portato al fatto che è diventato possibile prevedere teoricamente bersagli di successo per l'azione terapeutica, nonché modifiche delle strutture chimiche dei farmaci, fornendo nuovi composti con nuove proprietà. Pertanto, l'antibiotico sulfanilamide, a seguito di numerosi studi, ha dato origine a intere famiglie di farmaci ipoglicemizzanti, diuretici e antipertensivi. La progettazione dei farmaci ha raggiunto un livello qualitativamente nuovo quando lo sviluppo di nuovi composti farmaceutici è diventato non solo frutto dell'immaginazione dei chimici, ma il risultato di un dialogo scientifico tra biologi e chimici.
Una nuova svolta è stata associata allo sviluppo della biologia molecolare, che ha permesso di portare allo sviluppo informazioni sul genoma, clonare geni che codificano bersagli biologici terapeuticamente importanti ed esprimere i loro prodotti proteici.
Il completamento del progetto “genoma umano”, che ha segnato l'inizio del nuovo millennio, a seguito del quale è stata letta l'informazione completa contenuta nel DNA umano, è stato un vero trionfo per il ramo della scienza biologica chiamato “genomica”. La genomica fornisce un approccio completamente nuovo alla ricerca di nuovi bersagli terapeuticamente importanti, consentendo di ricercarli direttamente nel testo nucleotidico del genoma.
Il genoma umano contiene 12.000-14.000 geni che codificano per proteine secrete. Attualmente nell’industria farmaceutica non vengono utilizzati più di 500 target. Ci sono studi che affermano che molte malattie sono "multifattoriali", cioè sono causate dalla disfunzione non di una proteina o di un gene, ma di 5-10 proteine interconnesse e dei loro geni codificanti. Sulla base di queste considerazioni, possiamo concludere che il numero di target oggetto di studio dovrebbe aumentare di almeno 5 volte.
La classificazione biochimica dei bersagli biologici attualmente studiati e il loro rapporto numerico sono mostrati nella Figura 2. Va notato in particolare che la maggior parte (>60%) dei recettori sono recettori accoppiati a proteine G di membrana ( GPCR, Recettori accoppiati a proteine G), e il volume totale delle vendite di farmaci destinati a interagire con essi è pari a 65 miliardi di dollari all'anno e continua a crescere.
Concetti basilari

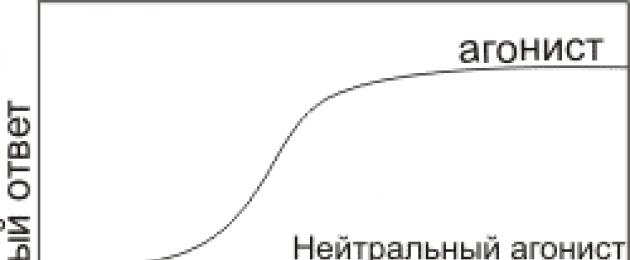
Figura 3. Tre tipi di influenza dei ligandi sulla risposta cellulare: aumento della risposta ( agonista positivo), costanza della risposta, ma competizione per il legame con altri ligandi ( agonista neutro) e risposta decrescente ( antagonista).
I concetti di base utilizzati nella progettazione della resistenza sono bersaglio E medicinale. Un bersaglio è una struttura biologica macromolecolare, presumibilmente associata a una determinata funzione, la cui violazione porta a una malattia e sulla quale deve essere esercitato un determinato effetto. Gli obiettivi più comuni sono i recettori e gli enzimi. Un farmaco è un composto chimico (solitamente a basso peso molecolare) che interagisce specificamente con il bersaglio e in un modo o nell'altro modifica la risposta cellulare creata dal bersaglio.
Se un recettore agisce come bersaglio, molto probabilmente il farmaco sarà il suo ligando, cioè un composto che interagisce specificamente con il sito attivo del recettore. In assenza di un ligando, il recettore è caratterizzato dal proprio livello di risposta cellulare, la cosiddetta attività basale.
In base al tipo di modificazione della risposta cellulare, i ligandi sono divisi in tre gruppi (Fig. 3):
- Gli agonisti aumentano la risposta cellulare.
- Gli agonisti neutri si legano al recettore ma non alterano la risposta cellulare rispetto al basale.
- Gli agonisti inversi, o antagonisti, diminuiscono la risposta cellulare.
Il grado di interazione del ligando con il bersaglio è misurato dall'affinità o dall'affinità. L'affinità è uguale alla concentrazione del ligando alla quale metà dei bersagli sono legati al ligando. La caratteristica biologica del ligando è la sua attività, cioè la concentrazione del ligando alla quale la risposta cellulare è pari alla metà del massimo.
Definizione e validazione del target
Uno dei primi e più importanti passi nella progettazione di un farmaco è la scelta del bersaglio giusto, che può essere manipolato in un modo specifico per regolare alcuni processi biochimici, lasciando gli altri il più inalterati possibile. Tuttavia, come già accennato, ciò non è sempre possibile: non tutte le malattie sono il risultato della disfunzione di una sola proteina o gene.
Con l'avvento dell'era post-genomica, il targeting avviene utilizzando metodi di genomica comparativa e funzionale. Sulla base dell'analisi filogenetica del genoma umano, vengono identificati i geni correlati a geni le cui funzioni dei prodotti proteici sono già noti e questi geni possono essere clonati per ulteriori ricerche.
Tuttavia, obiettivi le cui funzioni sono state determinate solo ipoteticamente non possono servire come punto di partenza per ulteriori ricerche. È necessaria una validazione sperimentale in più fasi, a seguito della quale è possibile comprendere la funzione biologica specifica del bersaglio in relazione alle manifestazioni fenotipiche della malattia in studio.
Esistono diversi metodi per la validazione del target sperimentale:
- i metodi genomici consistono nel sopprimere la sintesi del bersaglio nel sistema di test ottenendo mutanti genici knockout (in cui il gene bersaglio è semplicemente assente) o utilizzando sequenze antisenso di RNA che “spengono” l'uno o l'altro gene;
- i target possono essere inattivati con anticorpi monoclonali o irradiando il target modificato con cromoforo con luce laser;
- i bersagli possono essere inattivati con ligandi inibitori a basso peso molecolare;
- È anche possibile convalidare direttamente il bersaglio stabilendo la sua interazione con l'uno o l'altro composto mediante il metodo della risonanza plasmonica.
Il livello di validazione del target aumenta con il numero di animali modello (ceppi genetici speciali di animali da laboratorio) in cui la modificazione del target determina l'espressione fenotipica desiderata. Il più alto livello di validazione è, ovviamente, la dimostrazione che una modifica del bersaglio (ad esempio il blocco o l'eliminazione del recettore o l'inibizione dell'enzima) porta a sintomi clinicamente identificabili e riproducibili nell'uomo, tuttavia resta inteso che ciò può essere osservato abbastanza raramente.
Inoltre, quando si sceglie un bersaglio, non bisogna dimenticare un fenomeno come il polimorfismo, ovvero il fatto che un gene può esistere in diverse isoforme in diverse popolazioni o razze di persone, il che porterà a un diverso effetto del farmaco su pazienti diversi.
Una volta trovato e validato il bersaglio, inizia la ricerca diretta che dà come risultato numerose strutture di composti chimici, solo pochi dei quali destinati a diventare farmaci.
Lo studio di tutti i ligandi chimicamente possibili (“spazio chimico”) è impossibile: una semplice stima mostra che sono possibili almeno 10 40 diversi ligandi, mentre sono trascorsi solo ~ 10 17 secondi dalla creazione dell'universo. Pertanto, vengono imposte una serie di restrizioni sulla possibile struttura dei ligandi, che restringono significativamente lo spazio chimico (lasciandolo, tuttavia, completamente immenso). In particolare, per restringere lo spazio chimico, vengono imposte condizioni simili ai farmaci ( somiglianza con la droga), che in un caso semplice può essere espresso dalla regola del cinque di Lipinski, secondo la quale un composto, per “assomigliare” ad un farmaco, deve:
- avere meno di cinque atomi donatori di legame idrogeno;
- avere un peso molecolare inferiore a 500;
- avere una lipofilicità (log P - coefficiente di distribuzione della sostanza all'interfaccia acqua-ottanolo) inferiore a 5;
- avere un totale di non più di 10 atomi di azoto e ossigeno (una stima approssimativa del numero di accettori di legami idrogeno).
Come set iniziale di ligandi testati per la capacità di legarsi a un bersaglio, vengono solitamente utilizzate le cosiddette librerie di composti, fornite commercialmente da aziende specializzate in questo o contenute nell'arsenale di un'azienda farmaceutica che sviluppa un nuovo farmaco o lo ordina da una società terza. Tali librerie contengono migliaia e milioni di composti. Ciò, ovviamente, non è del tutto sufficiente per testare tutte le opzioni possibili, ma di norma non è necessario. Il compito in questa fase dello studio è identificare i composti che, dopo ulteriori modifiche, ottimizzazioni e test, possano fornire un "candidato" - un composto destinato alla sperimentazione sugli animali (studi preclinici) e sull'uomo (studi clinici).
Questa fase viene eseguita utilizzando lo screening ad alto rendimento ( in vitro) o il suo computer ( in silico) analisi - docking ad alte prestazioni.
Chimica combinatoria e screening ad alto rendimento
Lo screening è una procedura ottimizzata in pipeline, in seguito alla quale un gran numero di composti chimici (> 10.000) vengono testati per affinità o attività in relazione a uno speciale sistema di test (simulazione biologica). In base alle prestazioni, si distinguono diversi tipi di screening:
- bassa produttività (10.000–50.000 campioni);
- medio-produttivo (50.000-100.000 campioni);
- produttività elevata (oltre 100.000–5.000.000 di campioni).
Per lo screening, come per una procedura "industriale", l'efficienza, i costi e il tempo impiegato nell'operazione sono molto critici. Di norma, lo screening viene effettuato su installazioni robotiche in grado di funzionare 24 ore su 24 e tutto l'anno (Fig. 4).

Figura 4. Attrezzatura utilizzata per lo screening ad alta produttività. UN - Pipetta robotica, in modalità automatica ad alte prestazioni, che applica campioni dei composti testati in una piastra con un sistema di screening. Il numero tipico di depressioni su un dado è di migliaia. Il volume del sistema in un pozzo è di microlitri. Il volume del campione introdotto è di nanolitri. B - Impianto per lo screening e la lettura ad alte prestazioni del segnale fluorescente Mark II Scarina. Funziona con matrici contenenti 2048 rientranze (NanoCarrier). Completamente automatico (funziona 24 ore su 24). Produttività: più di 100.000 pozzi (campioni) al giorno.
Il principio dello screening è abbastanza semplice: nelle piastre contenenti un sistema di test (ad esempio, un bersaglio immobilizzato o cellule intere appositamente modificate), il robot estrae le sostanze da testare (o una miscela di sostanze) da una pipetta, seguendo un determinato programma. Inoltre, una piastra può contenere migliaia di "pozzetti" con un sistema di test e il volume di tale pozzetto può essere molto piccolo, così come il volume del campione introdotto (micro o addirittura nanolitri).
Quindi vengono letti i dati dalla piastra, indicando quale pozzo ha attività biologica e quale no. A seconda della tecnologia utilizzata, il rilevatore può leggere un segnale radioattivo, fluorescenza (se il sistema è costruito utilizzando proteine fluorescenti), bioluminescenza (se viene utilizzato un sistema luciferina-luciferasi o suoi analoghi), polarizzazione delle radiazioni e molti altri parametri.
Tipicamente, come risultato dello screening, il numero di composti testati viene ridotto di 3-4 ordini di grandezza. I composti per i quali durante il processo di screening è stata rilevata un'attività superiore a un determinato valore sono chiamati prototipi. Tuttavia, va capito che tale "buona fortuna" è ancora molto, molto lontana dalla cura finale. Solo quelli che mantengono la loro attività in sistemi modello e soddisfano una serie di criteri forniscono precursori di farmaci che vengono utilizzati per ulteriori ricerche.
Come già accennato, anche le librerie contenenti più di un milione di composti non sono in grado di rappresentare l'intero possibile spazio chimico dei ligandi. Pertanto, è possibile scegliere due diverse strategie di screening: screening diversificato e screening mirato. La differenza tra loro sta nella composizione delle librerie di composti utilizzati: nella variante di diversificazione vengono utilizzati ligandi quanto più diversi possibile tra loro in modo da coprire una regione quanto più ampia possibile dello spazio chimico, mentre nella variante quello focalizzato, al contrario, vengono utilizzate librerie di composti correlati ottenuti mediante chimica combinatoria, che consente, conoscendo la struttura approssimativa del ligando, di sceglierne la variante più ottimale. Il buon senso suggerisce che in un progetto su larga scala per creare un nuovo farmaco, entrambi questi approcci dovrebbero essere utilizzati in sequenza: in primo luogo, la diversificazione, al fine di identificare le classi più diverse di composti di successo, e poi focalizzati, al fine di ottimizzare la struttura di questi composti e ottenere prototipi funzionanti.
Se si conosce il bersaglio del cosiddetto spazio biologico, cioè le caratteristiche dei ligandi (dimensione, idrofobicità, ecc.) che possono legarsi ad esso, allora quando si compila la libreria dei composti testati, i ligandi che rientrano nel " intersezione” degli spazi biologici e chimici, poiché ciò ovviamente aumenta l’efficienza della procedura.
Le strutture prototipo risultanti dallo screening sono ulteriormente sottoposte a una serie di ottimizzazioni effettuate nella ricerca moderna, solitamente in stretta collaborazione tra diversi gruppi di ricercatori: biologi molecolari, farmacologi, modellatori e chimici medici (Fig. 5).

Figura 5. Ciclo farmacologico. Il gruppo di biologia molecolare è responsabile dell'ottenimento di target mutanti, il gruppo di farmacologia è responsabile della misurazione dei dati sull'attività e dell'affinità dei ligandi sintetizzati su target wild-type e mutanti, il gruppo di modellizzazione è responsabile della costruzione di modelli target, della previsione delle loro mutazioni e prevedendo le strutture dei ligandi, il gruppo di chimica farmaceutica è responsabile dei ligandi di sintesi.
Ad ogni giro di questo "ciclo farmacologico" il prototipo si avvicina al predecessore e poi al candidato, che viene già testato direttamente sugli animali (studi preclinici) e sull'uomo - nel corso degli studi clinici.
Pertanto, il ruolo dello screening è quello di ridurre significativamente (di diversi ordini di grandezza) il campione di prototipi (Fig. 6).

Figura 6. Il ruolo dello screening ad alto rendimento nello sviluppo di un nuovo farmaco. Screening, sia che si tratti di laboratorio ( in vitro) o computer ( in silico), è la procedura principale e più dispendiosa in termini di risorse per selezionare le strutture di partenza dei farmaci (prototipi) dalle librerie di composti disponibili. I risultati dello screening rappresentano spesso il punto di partenza per l’ulteriore processo di sviluppo del farmaco.
Ricerche cliniche
La medicina è un settore in cui non bisogna mai avere fretta. Soprattutto quando si tratta dello sviluppo di nuovi farmaci. Basti ricordare la storia del farmaco Talidamide, sviluppato alla fine degli anni '50 in Germania, il cui utilizzo da parte delle donne incinte portava alla nascita di bambini con malformazioni congenite degli arti, fino alla loro completa assenza. Questo effetto collaterale non è stato rilevato in tempo durante gli studi clinici a causa di test non sufficientemente approfonditi e accurati.
Pertanto, al momento, la procedura di test antidroga è piuttosto complicata, costosa e richiede molto tempo (2-7 anni di test in clinica e da 100 milioni di dollari per composto candidato, cm. riso. 7).

Figura 7. Il processo di sviluppo di un nuovo farmaco dura dai 5 ai 16 anni. Il costo dei test clinici di un singolo composto candidato è superiore a 100 milioni di dollari. Il costo totale dello sviluppo, compresi i farmaci che non hanno raggiunto il mercato, spesso supera il miliardo di dollari.
Innanzitutto, anche prima di entrare in clinica, i farmaci vengono esaminati per tossicità e cancerogenicità e dovrebbero essere condotti studi, ad eccezione dei sistemi in vitro su almeno due tipi di animali da laboratorio. I farmaci tossici, ovviamente, non entrano in clinica, tranne quando sono destinati al trattamento di malattie particolarmente gravi e non hanno ancora analoghi meno tossici.
Inoltre, i farmaci vengono sottoposti a studi di farmacocinetica, cioè vengono testate le caratteristiche fisiologiche e biochimiche come assorbimento, distribuzione, metabolismo ed escrezione (in inglese è abbreviato AGGIUNGIMI - Assorbimento, distribuzione, metabolismo ed estrazione). La biodisponibilità, ad esempio, è una sottocaratteristica dell'introduzione di un farmaco nell'organismo, che caratterizza il grado in cui perde le sue proprietà biologiche quando introdotto nell'organismo. Quindi, l'insulina assunta per via orale (per via orale) ha una bassa biodisponibilità, poiché, essendo una proteina, viene scomposta dagli enzimi gastrici. Pertanto, l'insulina viene somministrata per via sottocutanea o intramuscolare. Per lo stesso motivo vengono spesso sviluppati farmaci che agiscono in modo simile ai loro prototipi naturali, ma hanno una natura non proteica.
Dal punto di vista legale, il processo di sperimentazione clinica di nuovi farmaci presenta molte sfumature, poiché richiedono un'enorme quantità di documentazione di supporto (diverse migliaia di pagine in totale), permessi, certificazioni, ecc. Inoltre, molte procedure formali variano notevolmente da paese a paese a causa delle diverse legislazioni. Pertanto, per risolvere questi numerosi problemi, esistono aziende speciali che accettano ordini da grandi aziende farmaceutiche per condurre studi clinici e li reindirizzano a cliniche specifiche, accompagnando l'intero processo con una documentazione completa e assicurandosi che nessuna formalità venga violata.
Il ruolo dell’informatica nella progettazione dei farmaci
Attualmente, nella progettazione dei farmaci, come nella maggior parte degli altri settori ad alta intensità scientifica, il ruolo della tecnologia informatica continua ad aumentare. Va subito notato che l'attuale livello di sviluppo delle tecniche informatiche non consente lo sviluppo di un nuovo farmaco utilizzando solo i computer. I principali vantaggi forniti dai metodi computazionali in questo caso sono la riduzione del time to market per un nuovo farmaco e la riduzione dei costi di sviluppo.
Le principali tecniche informatiche utilizzate nella progettazione dei drag sono:
- modellazione molecolare (MM);
- proiezione virtuale;
- progettazione di nuovi farmaci de novo;
- valutazione delle proprietà di “somiglianza al farmaco”;
- modellazione del legame ligando-bersaglio.
Metodi MM basati sulla struttura del ligando
Se non si sa nulla della struttura tridimensionale del bersaglio (cosa che accade abbastanza spesso), si ricorre a metodi per creare nuovi composti basati su informazioni sulla struttura di ligandi già noti e dati sulla loro attività.
L'approccio si basa sul paradigma generalmente accettato in chimica e biologia, secondo il quale la struttura determina le proprietà. Sulla base dell'analisi delle correlazioni tra la struttura di composti noti e le loro proprietà, è possibile prevedere la struttura di un nuovo composto che presenta le proprietà desiderate (o, al contrario, prevedere le proprietà di una struttura nota). Inoltre, questo approccio viene utilizzato sia quando si modificano strutture note al fine di migliorarne le proprietà, sia quando si ricercano nuovi composti utilizzando lo screening di librerie di composti.
I metodi per determinare la somiglianza delle molecole (o metodi di fingerprinting) consistono nel prendere in considerazione in modo discreto alcune proprietà della molecola, chiamate descrittori (ad esempio, il numero di donatori di legami idrogeno, il numero di anelli benzenici, la presenza di un certo sostituente in una certa posizione, ecc.) e confrontando l'"impronta digitale" risultante con l'impronta di una molecola con proprietà note (usata come campione). Il grado di somiglianza è espresso dal coefficiente Tanimoto, che varia da 0 a 1. Un'elevata somiglianza implica la somiglianza delle proprietà delle molecole confrontate e viceversa.
I metodi basati sulle coordinate note degli atomi del ligando sono chiamati metodi di relazione quantitativa tra struttura e attività ( QSAR, Relazione quantitativa struttura-attività). Uno dei metodi più utilizzati di questo gruppo è il metodo di analisi comparativa dei campi molecolari ( COMFA, Analisi comparativa del campo molecolare). Questo metodo consiste nell'approssimare la struttura tridimensionale di un ligando mediante un insieme di campi molecolari che ne caratterizzano separatamente le proprietà steriche, elettrostatiche, donatore-accettrici e altre. Il modello CoMFA è costruito da un'analisi di regressione multipla di ligandi con attività nota e descrive un ligando che dovrebbe legarsi bene al bersaglio di interesse in termini di campi molecolari. L'insieme di campi risultante indica dove il ligando dovrebbe avere un sostituente voluminoso e dove dovrebbe essere piccolo, dove dovrebbe essere polare e dove non dovrebbe esserlo, dove dovrebbe essere un donatore di legami idrogeno e dove dovrebbe essere un accettore, e Presto.
Il modello può essere utilizzato nei compiti di screening virtuale delle librerie di composti, agendo in questo caso come un analogo di un farmacoforo. Lo svantaggio principale di questo metodo è che ha un elevato potere predittivo solo per classi di composti strettamente correlate; quando si tenta di prevedere l'attività di un composto di natura chimica diversa rispetto ai ligandi utilizzati per costruire il modello, il risultato potrebbe non essere sufficientemente affidabile.
Un diagramma di un possibile processo per la creazione di un nuovo farmaco basato sulla struttura del ligando è mostrato nella Figura 8.

Figura 8. Un esempio di modellazione molecolare basata sulla struttura del ligando. Per il peptide ciclico urotensina II ( in basso a sinistra) la struttura tridimensionale è stata determinata mediante spettroscopia NMR di una soluzione acquosa ( a sinistra in alto). La relazione spaziale dei residui aminoacidici del motivo TRP-LYS-TYR, importante per la funzione biologica, è stata utilizzata per costruire un modello farmacoforico ( in alto a destra). Come risultato dello screening virtuale, è stato scoperto un nuovo composto che dimostra attività biologica ( in basso a destra).
Ovviamente, l’affidabilità della modellazione, così come l’efficienza dell’intero processo di progettazione di un nuovo farmaco, può essere significativamente migliorata se si prendono in considerazione i dati non solo sulla struttura dei ligandi, ma anche sulla struttura della proteina bersaglio . I metodi che tengono conto di questi dati vengono collettivamente definiti progettazione della resistenza basata su informazioni strutturali ( SBDD, Progettazione di farmaci basata sulla struttura).
Metodi MM basati sulla struttura delle proteine
Grazie al crescente potenziale della biologia strutturale, è sempre più possibile stabilire la struttura tridimensionale sperimentale di un bersaglio, o costruire il suo modello molecolare basato sull'omologia con una proteina la cui struttura tridimensionale è già stata determinata.
I metodi più comunemente utilizzati per determinare la struttura tridimensionale delle biomacromolecole ad alta risoluzione (spesso, quando la struttura sperimentale del bersaglio non è ancora disponibile, si ricorre alla modellazione basata sull'omologia - un metodo per il quale viene dimostrato che il modello costruito da esso è di qualità sufficiente se l'omologia tra il modello strutturale e la proteina simulata non è inferiore al 40%.
La modellazione per omologia viene utilizzata particolarmente spesso nello sviluppo di farmaci che prendono di mira i recettori accoppiati a proteine G, poiché essi, essendo proteine di membrana, sono molto difficili da cristallizzare e proteine così grandi non sono ancora disponibili per l'NMR. Per questa famiglia di recettori è nota la struttura di una sola proteina, la rodopsina bovina, ottenuta nel 2000 a Stanford, che viene utilizzata come modello strutturale nella stragrande maggioranza degli studi.
Tipicamente, uno studio basato su dati strutturali prende in considerazione anche i dati sulla mutagenesi target per stabilire quali residui amminoacidici sono più importanti per la funzione proteica e il legame con i ligandi. Queste informazioni sono particolarmente preziose quando si ottimizza il modello costruito, che, essendo solo un derivato della struttura della proteina modello, non può tenere conto di tutte le specifiche biologiche dell'oggetto modellato.
La struttura tridimensionale del bersaglio, oltre a essere in grado di spiegare il meccanismo molecolare dell'interazione ligando-proteina, viene utilizzata nel docking molecolare o nella simulazione computerizzata dell'interazione ligando-proteina. Il docking utilizza come informazione di partenza la struttura tridimensionale della proteina (in questa fase dello sviluppo tecnologico, di regola, conformazionalmente immobile) e la struttura del ligando, la cui mobilità conformazionale e interposizione con il recettore è modellata nel modello processo di attracco. Il risultato del docking è la conformazione del ligando che meglio interagisce con il sito di legame della proteina, in termini di funzione di docking stimata, che si avvicina all'energia libera del legame del ligando. In realtà, a causa di molte approssimazioni, la funzione di valutazione non sempre correla con la corrispondente energia di legame sperimentale.
Il docking consente di risparmiare tempo e denaro eseguendo una procedura simile allo screening ad alto rendimento sui sistemi informatici. Questa procedura si chiama screening virtuale, e il suo vantaggio principale è che per i test farmacologici reali non è necessario acquistare un'intera libreria di un milione di composti, ma solo “prototipi virtuali”. Di solito, per evitare errori, lo screening e l'attracco vengono utilizzati contemporaneamente, completandosi a vicenda (Fig. 9).

Figura 9. Due opzioni per combinare lo screening ad alto rendimento e la modellazione molecolare. Sopra: screening iterativo sequenziale. In ogni fase della procedura viene utilizzato un insieme relativamente piccolo di ligandi; sulla base dei risultati dello screening viene costruito un modello che spiega la relazione tra struttura e attività. Il modello viene utilizzato per selezionare la serie successiva di ligandi da testare. Metter il fondo a: screening una tantum. Ad ogni passaggio, il modello viene costruito sul set di addestramento e utilizzato per fare previsioni sul set di test.
Con l'aumento della potenza del computer e l'emergere di algoritmi più corretti e fisici, il docking stimerà meglio l'energia di legame delle proteine al ligando, inizierà a tenere conto della mobilità delle catene proteiche e dell'influenza del solvente. Tuttavia, non è noto se lo screening virtuale sarà mai in grado di farlo completamente sostituire un vero esperimento biochimico; se è così, allora ciò richiede ovviamente un livello qualitativamente nuovo di algoritmi che attualmente non sono in grado di descrivere in modo assolutamente corretto l'interazione di un ligando con una proteina.
Uno dei fenomeni che illustrano l’imperfezione degli algoritmi di docking è il paradosso della somiglianza. Questo paradosso sta nel fatto che composti strutturalmente leggermente diversi possono avere attività radicalmente diverse e allo stesso tempo, dal punto di vista degli algoritmi di docking, essere praticamente indistinguibili.
I prototipi di farmaci possono essere ottenuti non solo scegliendo da un database di composti già preparato. Se esiste una struttura bersaglio (o almeno un modello farmacoforico tridimensionale), è possibile costruire ligandi de novo utilizzando i principi generali dell'interazione intermolecolare. Con questo approccio, uno o più frammenti molecolari di base vengono posizionati nel sito di legame del ligando e il ligando viene “costruito” in sequenza nel sito di legame, ottimizzato in ogni fase dell’algoritmo. Le strutture risultanti, proprio come nel docking, vengono valutate utilizzando funzioni di valutazione empirica.
Limitazioni all'uso dei metodi informatici
Nonostante tutte le loro promesse, i metodi informatici presentano una serie di limitazioni di cui è necessario tenere conto per immaginare correttamente le possibilità di questi metodi.
Innanzitutto, nonostante l'ideologia in silico implica la conduzione di esperimenti informatici a tutti gli effetti, cioè esperimenti i cui risultati sono di per sé preziosi e affidabili, è necessaria una verifica sperimentale obbligatoria dei risultati ottenuti. Cioè, è implicita una stretta collaborazione di gruppi scientifici che conducono un esperimento al computer con altri gruppi sperimentali (Fig. 5).
Inoltre, i metodi informatici non sono ancora in grado di tenere conto dell'intera varietà degli effetti di un farmaco sul corpo umano, per cui non sono in grado né di abolire né di ridurre significativamente i test clinici, che occupano la maggior parte del tempo nello sviluppo di un nuovo farmaco.
Pertanto, ad oggi, il ruolo dei metodi informatici nella progettazione dei farmaci si riduce all'accelerazione e alla riduzione dei costi della ricerca che precede gli studi clinici.
Trascina la prospettiva del design
Heinrich KLECH, direttore della ricerca e dello sviluppo medico, Centro medico regionale Eli Lilly, professore all'Università di Vienna:
1. Questo farmaco innovativo è un farmaco fondamentalmente nuovo che tratta la malattia con un meccanismo completamente diverso rispetto ai farmaci precedenti. Sono questi farmaci rivoluzionari ad avere successo commerciale nel mercato odierno. La medicina farmaceutica ha fatto passi da gigante negli ultimi anni.
I vecchi farmaci tradizionali, come l’aspirina, curavano solo i sintomi della malattia, e questa era l’era chimica dei prodotti farmaceutici. Negli ultimi anni, i ricercatori hanno iniziato a prestare molta più attenzione all'effetto dei composti biologici sui recettori, con i quali è possibile combattere davvero la causa della malattia. Quindi oggi trattano l'ipertensione, le malattie del cuore e del tratto gastrointestinale. I biopreparati hanno particolarmente successo nel trattamento del cancro.
La genetica si è unita alla moderna farmaceutica, studiando, tra le altre cose, le anomalie genetiche. Secondo loro, i farmacisti determinano qual è la reazione di un individuo umano a un particolare farmaco, sia classico che nuovo. In modo molto più specifico di prima, viene sviluppato un piano di trattamento per il paziente.
2. Esistono requisiti piuttosto severi per l'efficacia di un nuovo farmaco e la sua sicurezza. Inoltre, questi requisiti sono cambiati in modo significativo negli ultimi 20 anni. In precedenza, per ottenere una licenza, era sufficiente che le autorità di regolamentazione fornissero dati sulla conduzione di 2-3mila test o studi su un nuovo farmaco. Ora è necessario studiare il farmaco su 8-10mila persone. Per quanto riguarda la disponibilità di un farmaco moderno, in linea di principio dovrebbe essere massima. Ma è necessario anche un monitoraggio costante della sua assunzione da parte di un medico e l'acquisto (secondo la pratica occidentale consolidata) deve essere effettuato rigorosamente secondo la prescrizione.
3. La creazione di un nuovo farmaco richiede fino a 14 anni. Dipende dalla classe a cui appartiene il farmaco, da quanto i suoi "predecessori" sono conosciuti dal pubblico, ecc. La ricerca potrebbe richiedere da 500 milioni a un miliardo di dollari. Basti pensare che dei 100mila composti molecolari studiati, solo un migliaio possono diventare la base per un nuovo farmaco. Di queste, solo 100 molecole avranno un effetto attivo sul corpo del paziente. Ma anche tra questi, il 90% si rivela tossico, tanto che solo 10 composti iniziali vengono ampiamente venduti e solo tre hanno successo commerciale. Pertanto, le aziende farmaceutiche coinvolte nello sviluppo di nuovi farmaci investono dal 14 al 20% dei loro profitti nella ricerca.
4. Oggi è molto promettente sviluppare e promuovere prodotti farmacogenetici. Innanzitutto, non sono stati trattati prima. In secondo luogo, il trattamento di una serie di malattie, tra cui il morbo di Alzheimer, con i farmaci tradizionali non ha dato risultati positivi. Inoltre, i farmacisti di tutto il mondo devono accelerare lo sviluppo di farmaci contro il cancro. Si sono fatti alcuni progressi, ma le persone continuano a soffrire di malattie maligne, il che significa che dobbiamo continuare a cercare una panacea per loro. Una terza area di ricerca promettente è il diabete, poiché non esiste ancora un farmaco che affronti la causa alla base della malattia. Dopotutto, l'insulina estingue solo i suoi effetti.
Oleg SUPRYAGA, Direttore medico di Nycomed Russia-CIS, MD, Professore:
1. Per farmaco moderno si intende spesso un farmaco “alla moda”, un farmaco creato con l'ausilio delle nuove tecnologie. A mio parere, una medicina moderna è quella destinata a curare le malattie moderne (attualmente disponibili). La struttura delle malattie, così come la disponibilità di alcuni farmaci nelle diverse regioni economiche e geografiche del mondo, è diversa, quindi anche la frequenza di utilizzo dei diversi farmaci è diversa. Pertanto, la definizione di medicina moderna sarà diversa per ciascuna regione.
2. Deve soddisfare i criteri di qualità, sicurezza, accessibilità che la società può permettersi in relazione ai suoi membri. Di norma viene creato un organismo nazionale (pubblico o statale) al quale è delegata la funzione di controllo di qualità dei medicinali. Una società con un’economia ben sviluppata e costi sanitari elevati può attuare una regolamentazione non tariffaria limitando o chiudendo l’importazione di medicinali nel proprio territorio (mercato) da altri paesi economicamente meno sviluppati. In questo modo viene tutelata anche l’industria farmaceutica.
3. La gamma dei costi per la creazione di un nuovo farmaco va da 5 milioni di dollari USA a 1 miliardo di dollari USA o più. In diversi paesi in modi diversi, tutto dipende dai criteri dettati dalla società o dallo stato e che, a loro volta, sono determinati dal livello di sviluppo economico e tecnologico della società, in particolare dalla sua industria farmaceutica, dalla volontà della società , lo Stato o i singoli individui a spendere determinate altre somme di denaro per medicinali, medicinali e assistenza sanitaria.
4. La strategia di Nycomed è quella di esternalizzare il suo sviluppo preclinico di farmaci (divisione Ricerca e Sviluppo (R&S)) a un'altra società. Nycomed è attualmente coinvolta nello sviluppo di farmaci a partire dal livello di studi clinici. Nuove molecole promettenti che hanno superato con successo la fase degli studi preclinici e portate al livello di sperimentazioni cliniche sono concesse in licenza da aziende specializzate (biotecnologia, centri di ricerca, ecc.).
Allo stesso tempo, la società Nycomed, insieme agli studi clinici, porta il farmaco sul mercato (soprattutto europeo) e il suo supporto al marketing e alle vendite. Cardiologia, incl. interventistica, neurologia, endocrinologia, pediatria, reumatologia e altre aree della medicina.
Rustam IKSANOV, direttore del Centro per la ricerca scientifica e lo sviluppo (TsNIiR) dell'OAO Nizhpharm.
1. Oggi la medicina è considerata una merce, il che significa che è un elemento del mercato, esiste secondo le sue leggi.
2. Innanzitutto, un farmaco moderno deve avere una sicurezza ed un'efficacia ragionevoli e comprovate. Giustamente, le questioni relative alla qualità stanno guadagnando sempre più attenzione. All’estero esistono standard molto elevati che si applicano a tutte le fasi dello sviluppo di un nuovo farmaco, della ricerca e della sua produzione. Solo la rigorosa osservanza di tutte le norme e regole può garantire il rispetto delle proprietà attese ed effettive del farmaco.
Attualmente anche in Russia vengono introdotti attivamente standard internazionali di qualità. Un passo abbastanza serio in questa direzione sarà, spero, l'introduzione in Russia nel 2005 degli standard GMP (buone pratiche di produzione). Oggi, solo poche aziende soddisfano tali standard in un modo o nell’altro.
Un’altra questione importante è la disponibilità dei medicinali, che non può essere risolta senza l’intervento statale in questo settore. I pazienti devono avere la garanzia di un trattamento efficace e sicuro.
3. I nuovi farmaci hanno ancora molta strada da fare prima di arrivare sullo scaffale delle farmacie. È necessario non solo sviluppare un farmaco, ma anche condurre studi sugli animali, studi clinici e ottenere la registrazione statale del farmaco. Lo sviluppo di un farmaco fondamentalmente nuovo all’estero richiede circa 10 anni e costa circa mezzo milione di dollari. Sfortunatamente, non possedendo tali mezzi, oggi la Russia non è praticamente impegnata nello sviluppo di farmaci fondamentalmente nuovi.
Allo stesso tempo, va notato che esiste un potenziale scientifico per tale lavoro in Russia. Spero che riceva lo sviluppo necessario. Fondamentalmente, le aziende russe sono impegnate nello sviluppo di farmaci generici, i cosiddetti farmaci generici. Costa meno.
4. Senza un'analisi del mercato dei farmaci, senza tenere traccia delle tendenze attuali nello sviluppo degli standard di trattamento, è impossibile valutare correttamente le prospettive per lo sviluppo della farmacologia. Ad esempio, la nostra azienda utilizza attivamente una serie di ricerche di mercato e consultazioni di esperti leader per determinare le sue aree promettenti.
È difficile trovare una persona che non prenda medicine ad un certo punto della sua vita. E allo stesso tempo, è improbabile che molte persone pensino al fatto che in medicina, come nel fuoco di una lente, si concentrano i risultati delle scienze fondamentali: chimica organica e inorganica, fisiologia, biochimica, biofisica, ovviamente, farmacologia e un complesso di scienze farmaceutiche. Le conquiste di queste discipline fondamentali, grazie alla scienza delle sostanze medicinali, entrano in pratica e servono a beneficio dell'uomo. Pertanto, l'introduzione alla farmacologia, a cui è dedicato l'articolo, non ha solo un valore cognitivo, ma aiuta anche a studiare in modo più mirato le discipline biologiche e chimiche a scuola.
Il percorso del farmaco dal laboratorio al paziente
La creazione di un farmaco solitamente inizia nel laboratorio di un chimico organico o nel laboratorio di un fitochimico. Il primo crea composti ancora inesplorati, il secondo isola dalle piante singoli composti chimici o un gruppo di sostanze con struttura simile. Le sostanze create o isolate vengono poi trasmesse a un farmacologo che determina se le sostanze hanno l'effetto desiderato. Supponiamo che un farmacologo stia cercando sostanze che abbiano un effetto antipertensivo, ad es. abbassando la pressione sanguigna. Può andare in due modi. Il primo percorso si chiama selezione. Allo stesso tempo, il farmacologo spesso non sa, anche presumibilmente, quale struttura chimica dovrebbe avere un agente antipertensivo, e testa una sostanza dopo l'altra negli esperimenti sugli animali, selezionando quelle inefficaci (setaccio di screening). Questo è un metodo molto dispendioso in termini di tempo e spesso inefficace, ma a volte l'unico possibile, soprattutto quando si tratta di sviluppare gruppi di farmaci nuovi e sconosciuti. Lo screening viene utilizzato per trovare agenti antitumorali. Fu utilizzato per la prima volta all'inizio del secolo da P. Ehrlich per ottenere farmaci antisifilitici a base di composti organici dell'arsenico.
Il metodo più comunemente usato sintesi diretta. Il ricercatore accumula gradualmente materiale che mostra quali radicali chimici o altre strutture sono responsabili di questo o quel tipo di azione. Uno dei problemi principali della farmacologia è lo studio dei modelli struttura-azione. Si accumulano sempre più dati, sulla base dei quali vengono compilati programmi per computer. Già con un grado maggiore di probabilità è possibile prevedere la natura dell'azione del composto previsto per la sintesi e il successivo studio. L'esperimento è sempre decisivo, ma la conoscenza degli schemi generali di "struttura-azione" accorcia la strada verso il successo.
Supponiamo quindi che venga trovato un rimedio efficace in grado di provocare un effetto ipotensivo, ma il lavoro del farmacologo non finisce qui. Deve scoprire se il composto chimico ha proprietà tossiche che possono manifestarsi se usato come farmaco. Il farmacologo solitamente definisce la tossicità acuta, cioè. dose in grado di provocare la morte nel 50% degli animali da esperimento (LD 50 - dose letale); più bassa è questa dose, più tossica è la sostanza. Solo quella sostanza può diventare un medicinale la cui dose terapeutica (terapeutica) è significativamente (spesso 20 o più volte) inferiore alla LD 50 . L'intervallo di dosi dal minimo efficace al minimo tossico indica l'ampiezza dell'effetto terapeutico dei farmaci.
Il farmacologo determina anche la possibilità di effetti collaterali con la somministrazione prolungata del farmaco a dosi terapeutiche. La definizione di tossicità subcronica viene effettuata: il farmaco viene somministrato per un lungo periodo, spesso fino a 6 mesi o più. Allo stesso tempo, vengono determinate le funzioni di tutti i sistemi corporei, i parametri biochimici del sangue, un esame patoistologico degli organi degli animali da esperimento viene effettuato dopo la fine della somministrazione del farmaco. Questo studio consente di giudicare se il farmaco viola le funzioni degli organi e dei tessuti del corpo con una somministrazione prolungata, ad es. se la terapia a lungo termine con questo composto è sicura. Il farmacologo determina anche altri possibili effetti tossici del farmaco: il suo effetto sulla funzione riproduttiva (la capacità di produrre prole), l'effetto embriotossico (la capacità di influenzare l'embrione), l'effetto teratogeno (la capacità di causare deformità del feto), l'effetto mutageno effetto. Con l'aiuto di campioni speciali, studiano l'effetto del farmaco sull'immunità, la possibilità di un effetto cancerogeno del farmaco, la sua attività allergenica, ecc.
Allo stesso tempo, i farmacisti lavorano anche per determinare la forma di dosaggio più razionale. Questo conclude lo studio preclinico del farmaco. Ogni paese ha un'istituzione ufficiale che consente la sperimentazione clinica del farmaco e il suo successivo utilizzo come farmaco. In Russia, l'autorizzazione alla sperimentazione clinica del farmaco viene concessa dal Comitato Farmacologico del Ministero della Salute della Federazione Russa.
Un medico che ha ricevuto un farmaco da testare ha gli stessi compiti di un farmacologo, cioè: valutazione dell'effetto terapeutico del farmaco e delucidazione della possibilità di effetti collaterali durante il suo utilizzo. Tuttavia, il clinico si trova ad affrontare difficoltà che il farmacologo sperimentale non affronta: la coscienza della persona che assume il farmaco può modificare la valutazione dell'effetto del farmaco. In alcune malattie, è possibile migliorare le condizioni del paziente sotto l'influenza del suggerimento e dell'autorità del medico, nonché del regime ospedaliero e della dieta, che hanno un effetto positivo. Pertanto, è necessario distinguere il vero effetto del farmaco dall'influenza di fattori concomitanti. A tale scopo viene utilizzato un test placebo (fittizio). Supponiamo che a un gruppo di pazienti, ovviamente, che non necessitano di un trattamento urgente ed efficace, vengano prescritte pillole contenenti un farmaco e all'altro gruppo vengano prescritte pillole che sono simili nell'aspetto, ma non contengono farmaci, un placebo. Se, allo stesso tempo, a seguito del trattamento, lo stato di salute migliora in circa il 60% dei pazienti del primo gruppo e nel secondo gruppo nel 30% dei pazienti, allora si verifica un significativo eccesso di effetto del farmaco rispetto al placebo. Pertanto, il farmaco è efficace. Se l'effetto del farmaco è uguale al placebo, si dovrebbe riconoscere l'inefficacia del farmaco. Lo sviluppo del farmaco è impegnato in una disciplina relativamente giovane: la farmacologia clinica. Se a seguito di studi clinici viene dimostrato che il farmaco è efficace, il medico deve comunque valutare la possibilità di effetti collaterali: gli effetti indesiderati dei farmaci. Se, ad esempio, un medico usa un farmaco per abbassare la pressione sanguigna e allo stesso tempo osserva un disturbo intestinale in un paziente durante il trattamento con un farmaco antipertensivo, allora questo è un esempio di effetto collaterale. Il grado e la gravità degli effetti collaterali sono tali da costringerci a rifiutarci di testare il farmaco e quindi a interrompere l'ulteriore sviluppo del farmaco. Tuttavia, un effetto collaterale leggermente pronunciato che non rappresenta una minaccia immediata per la salute del paziente non serve come motivo per rifiutare il farmaco. È noto che i diuretici, come furosemide, diclotiazide, riducono la concentrazione di potassio nel sangue, cioè. causare ipokaliemia. Tuttavia, tale violazione viene corretta prescrivendo una dieta ricca di questi ioni, oppure prescrivendo integratori di potassio o altri cosiddetti diuretici risparmiatori di potassio. La correzione consente di trattare con successo pazienti con malattie cardiovascolari con diuretici, senza preoccuparsi dello sviluppo di ipokaliemia.
Se gli studi clinici hanno successo, il farmaco riceve l’autorizzazione per la produzione e l’uso industriale ed entra nella rete delle farmacie. Recensioni a riguardo sono pubblicate sulla stampa, il meccanismo della sua azione continua a essere studiato e, infine, il farmaco prende il posto che gli spetta nell'arsenale dei medicinali. Il percorso di un nuovo farmaco dalla prima fase di ricerca al paziente è complicato e lungo. Molto spesso, sono necessari diversi anni prima che il farmaco possa essere utilizzato nella pratica. Delle molte migliaia di composti studiati, solo pochi vengono introdotti nella pratica e ricevono questo nome medicinale anche se ovviamente ci sono altri esempi.
Problemi di farmacocinetica
La farmacocinetica è una branca della farmacologia che studia il comportamento dei farmaci nell'organismo: il loro assorbimento, distribuzione, escrezione e biotrasformazione. Perché un farmaco funzioni, deve essere introdotto nel corpo. Tutte le vie di somministrazione si dividono in due gruppi: enterale e parenterale (dal greco. enterone- tratto gastrointestinale). Le vie di somministrazione enterale comprendono l'introduzione attraverso la bocca (anche sotto la lingua), nel duodeno e nel retto. Le vie di somministrazione parenterale che bypassano il tratto gastrointestinale comprendono la somministrazione sottocutanea, intramuscolare ed endovenosa di farmaci. La via di somministrazione determina in gran parte la velocità di ingresso e la gravità dell’effetto del farmaco.
Dopo essere stata introdotta nell'organismo, la sostanza medicinale viene trasportata dal sangue agli organi, ai tessuti e ai mezzi liquidi, ma ciò non significa che la concentrazione del farmaco somministrato in ciascun organo o tessuto sia la stessa. La distribuzione uniforme del farmaco è impedita dalle barriere tissutali attraverso le quali le sostanze medicinali penetrano in modo tutt'altro che uniforme. Una di queste barriere è la barriera ematoencefalica: la penetrazione di sostanze dal sangue nel sistema nervoso centrale è limitata, poiché le sostanze ionizzate o lipoinsolubili non penetrano nel cervello attraverso questa barriera. Ad esempio, le sostanze contenenti un atomo di azoto quaternario non penetrano bene attraverso questa barriera; tali sostanze possono includere il composto biologicamente attivo acetilcolina. Il significato biologico di una tale barriera è evidente: la penetrazione di determinate sostanze dal sangue nel cervello ne comprometterebbe notevolmente la funzione. Pertanto, non solo le sostanze biologicamente attive, ma anche molte sostanze medicinali (rilassanti muscolari, bloccanti gangliari) non penetrano nella barriera ematoencefalica.
Una barriera molto più permeabile è la parete capillare, attraverso la quale la maggior parte delle sostanze medicinali penetra nei tessuti, ma non passano le sostanze ad alto peso molecolare, ad esempio la proteina dell'albumina, che ha un peso molecolare di circa 70.000. in pratica: ad esempio, un gruppo di sostanze ad alto peso molecolare (le poliglucine) vengono utilizzate come sostituti del sangue, poiché circolano nel sangue senza penetrare nei tessuti. Anche la barriera placentare che separa il corpo della madre dal feto è facilmente permeabile ai farmaci. Pertanto, i farmaci somministrati al corpo materno possono avere un effetto anche sul feto, di cui bisogna tener conto quando si trattano le donne in gravidanza.
Le sostanze medicinali, soprattutto quelle solubili in acqua, vengono escrete dal corpo attraverso i reni. Le sostanze volatili vengono escrete dai polmoni, in parte i composti possono essere escreti con le feci e anche con le ghiandole sudoripare. Il rilascio di farmaci è uno dei motivi per cui la concentrazione del farmaco nel sangue diminuisce e l'efficacia della sua azione diminuisce.
Inoltre, i farmaci subiscono processi di biotrasformazione. La maggior parte dei farmaci sono liposolubili e sono acidi o basi organici deboli che vengono escreti relativamente poco dal corpo. Ad esempio, dopo la filtrazione nei glomeruli renali, vengono riassorbiti per diffusione attraverso le membrane e le giunzioni intercellulari delle cellule dei tubuli renali. Per una rapida eliminazione, i farmaci devono essere convertiti in forme più polari. Pertanto, se nel processo di biotrasformazione nell'organismo si formano metaboliti più polari, ionizzati a pH fisiologico, meno legati alle proteine plasmatiche, alle proteine tissutali, hanno meno capacità di penetrare attraverso le membrane del tubulo renale. Pertanto non vengono riassorbiti nei tubuli renali e vengono escreti nelle urine. Questo è ciò a cui servono i processi di biotrasformazione nel corpo, che contribuiscono all'escrezione del farmaco e lo rendono meno attivo.
Le reazioni chimiche coinvolte nella biotrasformazione si dividono in reazioni di sintesi (coniugazione) e reazioni non sintetiche. I primi comprendono reazioni di aggiunta di prodotti metabolici a sostanze medicinali. Sono note reazioni di acetilazione, ad es. aggiunta di residui di acido acetico, glucuronico e solforico. Le reazioni di sintesi coinvolgono anche gruppi sulfidrilici, che legano molti composti organici e inorganici, in particolare metalli pesanti. Le reazioni non specifiche includono reazioni di ossidazione, riduzione e idrolisi.
I sistemi enzimatici coinvolti nella biotrasformazione sono localizzati nel fegato e nel reticolo endoplasmatico delle cellule epatiche. Identificati nell'esperimento, vengono chiamati enzimi microsomiali, poiché sono associati alla frazione di microsomi rilasciati durante la centrifugazione differenziale di frammenti di cellule epatiche. Gli enzimi microsomiali catalizzano le reazioni di coniugazione e ossidazione, mentre le reazioni di riduzione e idrolisi sono spesso catalizzate da enzimi non microsomiali.
L'attività degli enzimi microsomiali varia da persona a persona ed è geneticamente determinata; dipende dalle caratteristiche genetiche dell'organismo. Si ritiene che l'entità della biotrasformazione negli individui possa variare di 6 volte o più, il che determina la sensibilità individuale al farmaco. Pertanto, in alcuni pazienti, l'effetto desiderato può essere ottenuto con dosi molte volte superiori rispetto ad altri e viceversa. Alcuni farmaci aumentano l'attività degli enzimi microsomiali, vengono chiamati induttori, altro - inibitori - sopprimerli.
Un esempio dell'importanza dell'attività degli enzimi microsomiali nella terapia è il farmaco antitubercolare isoniazide. Alcuni pazienti hanno un'elevata attività degli enzimi microsomiali, come li chiamano inattivatori rapidi dell'isoniazide, in altri pazienti questa attività è bassa, vengono chiamati inattivatori lenti. Dopo una somministrazione del farmaco per sei giorni in pazienti con bassa attività, la concentrazione di isoniazide nel sangue è 2,5 volte superiore rispetto al primo. Negli inattivatori lenti la dose deve essere ridotta per evitare effetti collaterali indesiderati del farmaco.
Naturalmente, i farmaci "biotrasformano" non solo il fegato, ma anche altri tessuti. Come risultato della biotrasformazione, le sostanze medicinali vengono convertite in metaboliti, che, di regola, sono meno attivi della sostanza principale, sono meglio solubili e vengono escreti relativamente facilmente dal corpo attraverso i reni. Pertanto, il corpo viene liberato dal farmaco somministrato.
La farmacocinetica implica la determinazione del tasso di inattivazione e rilascio, entrambi i processi sono definiti dal termine quota di eliminazione. Determina la percentuale di una sostanza proveniente da una dose somministrata che viene metabolizzata ed escreta durante la giornata. Se questa percentuale è piccola, il farmaco può accumularsi nell'organismo durante le dosi successive e aumentarne l'effetto. Il medico può sfruttare abilmente questo fenomeno scegliendo una dose di farmaco che satura l’organismo, per poi passare ad una dose più bassa che compensa la perdita del farmaco e si chiama dose di mantenimento. Alcune sostanze, come i glicosidi della digitale, vengono utilizzate in questo modo.
Continua
Il processo inizia con l'ottenimento di una nuova composizione chimica. Le sostanze con struttura complessa possono essere ottenute da varie fonti, come piante (glicosidi cardiaci), tessuti animali (eparina), colture microbiche (penicillina), cellule umane (urochinasi), ingegneria genetica (insulina umana). Una persona penetra più in profondità nelle relazioni strutturali e funzionali, la ricerca di nuovi agenti diventa più mirata.
Test preclinici
I test preclinici raccolgono informazioni sugli effetti biologici delle nuove sostanze. Lo screening iniziale viene effettuato in studi biochimico-farmacologici o esperimenti su colture cellulari, cellule isolate e organi isolati. Poiché questi modelli non sono in grado di riprodurre completamente l’intero complesso dei processi biologici in un organismo intatto, qualsiasi potenziale farmaco deve essere testato sugli animali. Solo gli esperimenti sugli animali possono rispondere alla domanda. se gli effetti desiderati si verificano a dosi non tossiche o poco tossiche.
Lo studio di tossicità è progettato per valutare:
- tossicità con l'uso a breve e lungo termine,
- la possibilità di danno genetico (genotossicità, mutagenicità),
- la possibilità di sviluppare tumori (onco- e cancerogenicità),
- la possibilità della nascita di un feto malato (teratogenicità).
Negli animali, i composti da testare vengono anche testati per l'assorbimento, la distribuzione, il metabolismo e l'escrezione (farmacocinetica). Anche a livello di studi preclinici, la stragrande maggioranza dei potenziali composti farmaceutici viene eliminata e ne rimangono solo pochi.
Test clinici
Fase I
In questa fase viene effettuato uno studio di nuovi farmaci in individui sani al fine di determinare se gli effetti riscontrati nei test sugli animali si osservano nell'uomo, per identificare la relazione tra dose ed effetto.
Fase II
Un potenziale nuovo farmaco è in fase di sperimentazione su pazienti selezionati per determinarne l'efficacia terapeutica nella malattia a cui è destinato. L'effetto positivo deve essere chiaro e gli effetti indesiderati sono accettabilmente piccoli.
Fase III
In questa fase vengono reclutati nello studio ampi gruppi di pazienti, con i quali il farmaco in studio viene confrontato con il trattamento standard per i risultati della terapia.
In quanto forma di sperimentazione umana, tali studi clinici sono soggetti a revisione e approvazione da parte di comitati etici in conformità con le Dichiarazioni di Helsinki, Tokyo e Venezia. Nel corso degli studi clinici, molti nuovi farmaci risultano inadatti all’uso. Alla fine, delle circa 10.000 sostanze appena ottenute, rimane solo un farmaco.
La decisione di approvare un nuovo farmaco viene presa dall'organismo nazionale di regolamentazione (in Russia, il Comitato Farmaceutico del Ministero della Salute della Federazione Russa). I richiedenti (aziende farmaceutiche) presentano all'autorità di regolamentazione una serie completa di documentazione di studi preclinici e clinici in cui i dati ottenuti sull'efficacia e la sicurezza soddisfano i requisiti stabiliti e la forma prevista per il rilascio del prodotto (compresse, capsule, ecc.)
Una volta approvato, un nuovo farmaco può essere commercializzato con il marchio e quindi. diventa disponibile per la prescrizione da parte dei medici e la vendita nelle farmacie.
Parallelamente è in corso lo sviluppo di un processo tecnologico per la produzione di un medicinale, requisiti di qualità e metodi di analisi.
Il processo di sviluppo dei farmaci e di preparazione alla produzione dura solitamente dai 5 agli 8 anni.
Fase IV
Man mano che il farmaco si diffonde, continua a essere monitorato. Il giudizio finale sul rapporto rischi/benefici di un nuovo farmaco può essere espresso solo sulla base di un’esperienza a lungo termine con il suo utilizzo. Pertanto, viene determinato il valore terapeutico di un nuovo farmaco.
La nostra opinione
Il percorso di un nuovo farmaco dal laboratorio di ricerca al banco della farmacia è lungo e richiede ingenti investimenti. Ecco perché è stupido parlare di sostituzione totale delle importazioni nell’industria farmaceutica. A meno che, ovviamente, non si tratti di copia illegale e semi-legale degli sviluppi di altri o della produzione infinita di farmaci obsoleti.
Come vengono creati i farmaci?
XIX secolo - inizio XX secolo
Modi per creare farmaci |
|
Isolamento di estratti da piante medicinali | Ricerca delle proprietà medicinali nelle sostanze inorganiche |
Visita medica |
|
Sugli animali - per tossicità | Nell'uomo - per la presenza di proprietà medicinali |
Ci sono molti farmaci tossici e inefficaci nelle farmacie, molti farmaci funzionano grazie all'effetto placebo. I ritrovamenti riusciti sono rari. |
|
Il fatto che la corteccia del salice potesse alleviare il calore e il dolore era noto anche ai guaritori. Ma ufficialmente i medici europei non lo usavano. Importavano il chinino dall'estero, che veniva usato per curare la febbre.
Questo finché la politica non è intervenuta nella storia degli antidolorifici e degli antipiretici.
Napoleone stabilì un blocco economico per l'Inghilterra e chiuse la terraferma alle navi mercantili inglesi. Per questo motivo il chinino smise di scorrere e si ricordarono del salice. E da esso è stato ottenuto abbastanza rapidamente l'acido salicilico.
Ma ahimè... Questo acido nella sua forma pura aveva un sapore sgradevole, provocava nausea, vomito ed era causa di forti dolori allo stomaco.
Molti medici hanno cercato di migliorare la tolleranza dell'acido salicilico, pur mantenendo le sue eccellenti proprietà. Ma solo il chimico tedesco Felix Hoffmann ci riuscì.
Suo padre soffriva di dolori lancinanti a causa di reumatismi cronici e riusciva a malapena a muoversi. Volendo alleviare la sofferenza di suo padre, Hoffman Jr. iniziò a lavorare per migliorare l'acido salicilico.
Ha elaborato la sostanza naturale in vari modi allora conosciuti. L'acido acetilsalicilico è stata la modifica di maggior successo. Rilasciato poi sotto il nome di "aspirina", divenne uno dei medicinali più famosi al mondo. È curioso che il meccanismo d'azione dell'aspirina sia stato scoperto solo dopo 100 anni di utilizzo.
Metà del XX secolo
Modi per creare farmaci |
||
Ricerca delle proprietà medicinali nelle sostanze inorganiche e organiche | ||
Visita medica |
||
Sugli animali - Per la tossicità e la presenza di proprietà medicinali, modellazione delle malattie umane nei rappresentanti della fauna | Sulle persone - Per le proprietà medicinali | Sulle colonie di microrganismi - Per rilevare le proprietà antimicrobiche |
L'avvento degli antibiotici e dell'insulina. In farmacia si trovano farmaci sempre più efficaci, ma gli effetti collaterali di molti farmaci sono ancora molto elevati. La modificazione chimica di centinaia e migliaia di composti porta alla scoperta dei sulfamidici, dei diuretici, degli ipoglicemizzanti e dei primi antipertensivi. Le vitamine sono incluse nella pratica. |
||
Le medicine più famose che sono arrivate fino a noi
È stato scoperto dal chirurgo canadese Fred Banting. Ha studiato le proprietà degli estratti del pancreas sugli animali. Quale fu la sua sorpresa quando, dopo l'introduzione di un simile estratto, un cane sopravvisse, morendo di diabete. Lo scienziato ha suggerito che alcune sostanze del pancreas abbassano i livelli di zucchero nel sangue. E dopo un po' testò la sua scoperta su un amico medico che soffriva di diabete.
Il nuovo farmaco ha causato un'ondata di energia e vigore in un amico malato.
E i test hanno mostrato una diminuzione della glicemia. Da allora, l’insulina è stata lo strumento principale nella lotta contro il diabete mellito grave.
Penicillina
L'antibiotico penicillina fu scoperto nel 1929 dal microbiologo inglese Alexander Fleming. Una volta, mentre studiava le proprietà degli stafilococchi, dimenticò una tazza con una coltura di batteri sul tavolo del laboratorio.
Tornando, lo scienziato ha trovato della muffa nella tazza. Con sua sorpresa, ha soppresso la crescita dei microbi. Il ricercatore ha avuto un'intuizione: la muffa rilascia una sostanza che uccide i batteri.
Ha chiamato questa sostanza "in onore" del fungo penicillium con cui ha lavorato. Test sugli animali hanno dimostrato che la penicillina è effettivamente efficace nell’uccidere i germi. E quando introdotto nel sangue non danneggia il corpo.
Il primo utilizzo efficace della penicillina è avvenuto in America. Il medicinale ha salvato la vita ad una giovane donna, madre di tre figli. Ha avuto una temperatura superiore a 40°C per 11 giorni, ed è morta lentamente. Ma il farmaco miracoloso la riportò alla coscienza già il secondo giorno di utilizzo. La donna sopravvisse e visse fino a tarda età.
Da allora, la penicillina ha salvato milioni di persone in tutto il mondo. E continua ad essere utilizzato fino ad oggi.
Fine del XX secolo - XXI secolo
Modi per creare farmaci |
|||
Isolamento, modificazione chimica di estratti di piante medicinali ed estratti dal corpo di animali | Ampia applicazione della simulazione al computer | Ricerca mirata di composti con proprietà medicinali basata sulle conoscenze di biochimica, fisiologia e genetica | Sintesi su larga scala di composti organici e ricerca delle loro proprietà medicinali |
Modi per verificare |
|||
Sugli animali - per proprietà medicinali, tossicità, capacità di provocare mutazioni, deformità fetali e cancro | Nell'uomo - per la presenza di proprietà medicinali. Lo studio dell'assorbimento, delle trasformazioni e delle vie di escrezione dei farmaci | Su colonie di microrganismi - per rilevare proprietà antimicrobiche | Sul computer - Per abbinare la molecola del farmaco bersaglio nel corpo |
L'emergere di migliaia di farmaci efficaci e relativamente sicuri. |
|||
- In contatto con 0
- Google Plus 0
- OK 0
- Facebook 0






