Scopo della lezione: Studiare le caratteristiche cliniche generali delle malattie autosomiche e gonosomiche. Determinare le principali indicazioni per la nomina di un esame citogenetico nei bambini e negli adulti. Impara a leggere un cariotipo.
Piano di lezione:
1. Definizione del termine “Malattia cromosomica”.
2. Cariotipo normale, sue caratteristiche. cromosomi omologhi.
3. Eziologia delle malattie cromosomiche: mutazioni genomiche e cromosomiche.
4. Regole generali per la registrazione delle anomalie cromosomiche e la lettura del cariotipo.
5. Caratteristiche cliniche generali delle malattie autosomiche. Le malattie più comuni.
6. Caratteristiche cliniche generali delle malattie gonosomiche.
7. Anomalie cromosomiche in individui clinicamente sani.
8. Indicazioni per la nomina di un esame citogenetico.
Termini di base:
Cariotipo - numero e insieme di caratteristiche morfologiche di un set cromosomico umano completo .
Idiogramma - Una rappresentazione schematica della striatura differenziale di un cromosoma, che consente di determinare la correttezza della sua struttura.
Cromosomi omologhi - i cromosomi accoppiati che contengono lo stesso insieme di geni sono morfologicamente simili, si coniugano durante la meiosi e possono scambiarsi regioni durante l'incrocio. A differenza dei cromatidi fratelli sintetizzati sulla base di una singola molecola di DNA, i cromosomi omologhi non sono identici nella composizione nucleotidica e possono portare diverse forme alleliche dello stesso gene.
Mutazioni genomiche - anomalie numeriche deviazioni dal numero normale di cromosomi (46).
Poliploidia - cambiamenti nel numero di cromosomi, multipli del set aploide:
Aneuploidia - cambiamenti nel numero di cromosomi che non sono multipli del set aploide, di regola, all'interno di una coppia di omologhi.
Riarrangiamenti strutturali dei cromosomi (mutazioni cromosomiche) - violazioni della struttura dei cromosomi, che portano o meno allo squilibrio cromosomico.
Mosaicismo - la presenza simultanea nel corpo di più cloni con cariotipi diversi.
Trisomia- la presenza di un terzo cromosoma omologo.
Monosomia- l'assenza di uno degli omologhi.
Polisomia- un aumento del numero di cromosomi all'interno di una coppia di omologhi
autosomi Tutti i cromosomi di un organismo tranne i cromosomi sessuali.
gonosomi-cromosomi sessuali.
Principali classificazioni:
- Mutazioni genomiche (anomalie numeriche)
- poliploidia nell'uomo si riscontra la triploidia di 69 cromosomi (3n) - che porta all'aborto o alla morte del bambino subito dopo la nascita, la tetraploidia -92 cromosomi (4n) - letale, che porta all'aborto precoce.
- aneuploidia
- monosomia l'assenza di uno di una coppia di cromosomi omologhi. L'unica forma conosciuta è 45,X.
- Trisomia - la presenza di un terzo cromosoma omologo. La causa delle malattie cromosomiche più comuni. Sindrome di Down - trisomia 21, sindrome di Patau - trisomia 13, sindrome di Edwards - trisomia 18.
- Polisomia (tetrasomia , pentasomia ) - la presenza di diversi (quattro, cinque) cromosomi omologhi. Nei nati vivi è stata descritta solo per i cromosomi sessuali. Ad esempio, 48,XXXX.
Per designare anomalie numeriche degli autosomi durante la registrazione di un cariotipo, viene utilizzato il seguente approccio: 1) un numero che indica il numero totale di cromosomi, 2) la formula per l'insieme dei cromosomi sessuali. 3) segni (+) o (-), seguiti dal numero di cromosomi persi o in eccesso. Ragazzo con sindrome di Down 47, XY, +21
- Mutazioni cromosomiche (riarrangiamenti strutturali)
- Equilibrato e
- Inversioni (es nv) rotazione di una sezione di un cromosoma di 180 gradi senza perdita di materiale genetico.
- Traslocazioni(T)- scambio di materiale genetico tra cromosomi
- Traslocazioni Robertsoniane (rob) – unione di due cromosomi acrocentrici in un'unica struttura morfologica
- Squilibrato
- Eliminazioni(del)– perdita di materiale genetico
- Duplicazioni(dup)- un cambiamento in un cromosoma in cui una delle sue regioni è rappresentata linearmente due o più volte.
- Isocromosomi(i) Cromosomi costituiti da due bracci corti o due lunghi con contemporanea perdita di materiale dall'altro braccio.
I riarrangiamenti bilanciati non portano alla comparsa di malattie cromosomiche nei portatori, ma possono causare infertilità, aborti ricorrenti o la nascita di bambini con malattie cromosomiche. I riarrangiamenti sbilanciati sono la causa delle malattie cromosomiche.
La registrazione di una riorganizzazione strutturale in un cariotipo inizia sempre con un'abbreviazione di una o tre lettere, quindi molto spesso i cromosomi e i punti di interruzione coinvolti sono elencati tra parentesi. Ad esempio: 46,XX,ins(7;2)(p13;q22q32). Secondo la nomenclatura internazionale, le abbreviazioni degli aggiustamenti strutturali sono le seguenti: inv- inversione, T- traslocazione reciproca, ins- traslocazione o inserzione non reciproca, rapinare– Traslocazione Robertsoniana, del- cancellazione R- cromosoma circolare dup- duplicazione io- isocromosoma.
L'ordine di registrazione delle anomalie strutturali. Dopo il numero di cromosomi e l'insieme dei cromosomi sessuali, denominare il riarrangiamento utilizzando simboli standard, quindi tra parentesi i cromosomi coinvolti nel riarrangiamento. Prima il sesso, poi gli autosomi. Sono separati (;) Nelle parentesi seguenti vengono separati anche i punti di interruzione (;).
Classificazione delle malattie cromosomiche. Clinicamente suddiviso autosomico malattie e disturbi nel sistema dei cromosomi sessuali ( gonosomiale malattie).
Malattie autosomiche - trisomie complete, trisomie a mosaico. Adeguamenti strutturali che si sono verificati de novo o come risultato di riarrangiamenti genitoriali equilibrati, clinicamente indicati trisomia parziale O monosomia parziale.
Malattie gonosomiche monosomia, trisomia, polisomia, alterazioni strutturali.
Schemi e disegni di base:
Cariogramma di un corredo cromosomico normale. A sinistra di ciascun cromosoma c'è il suo idiogramma.
Descrizioni di base:
Nonostante il fatto che malattie cromosomiche come la sindrome di Down, la sindrome di Edwards, alcune delle sindromi più comuni di trisomia parziale e monosomia abbiano un quadro clinico caratteristico, la diagnosi finale viene fatta solo dopo il cariotipo. Inoltre, molti casi di cariotipo sbilanciato con un leggero squilibrio cromosomico non forniscono un quadro chiaro della sindrome cromosomica. Pertanto, prima di tutto, è necessario capire in quali casi è necessario l'invio per il cariotipo.
malattia autosomica dovrebbe essere sospettato nel caso di una combinazione di tre situazioni in un paziente:
a) Malformazioni congenite multiple, es. la presenza di almeno due malformazioni congenite patogeneticamente indipendenti tra loro e che colpiscono almeno due apparati.
b) Molteplici anomalie esterne e microanomalie dello sviluppo
c) Ritardo nello sviluppo psicomotorio del bambino o compromissione dell'intelletto del bambino.
La combinazione di queste condizioni si verifica in tutti i casi di malattie cromosomiche, le più comuni delle quali sono:
Sindrome di Down. Esistono tre forme citogenetiche della sindrome di Down: trisomia regolare (93% dei casi), traslocazione (5%) e mosaico (2%). Le principali caratteristiche cliniche della sindrome di Down sono ritardo mentale, ipotensione muscolare, incisione epicanto e mongoloide delle rime palpebrali, strabismo, labbra ispessite, lingua ispessita e solcata (la cosiddetta "lingua geografica o piegata"), dorso piatto della naso, palato stretto, padiglioni auricolari deformati, pelle in eccesso sul collo, linea trasversale del palmo (la cosiddetta “piega della scimmia”), clinodattilia dei mignoli. Tra le anomalie degli organi interni si notano difetti cardiaci (difetti dei setti in combinazione con anomalie dei grandi vasi), tratto gastrointestinale, sistema urinario e cervello. I bambini con sindrome di Down soffrono di un profondo ritardo mentale fino all'imbecillità. I singoli tipi di attività mentale soffrono a vari livelli.
Sindrome di Patau. Sindrome della trisomia 13. Nella maggior parte dei casi si presenta come una trisomia regolare, i riarrangiamenti strutturali, solitamente sotto forma di traslocazioni robertsoniane, sono descritti solo in un piccolo numero di casi. I bambini con la sindrome di Patau nascono con ipoplasia prenatale (peso corporeo fino a 2500 g). I principali segni della sindrome di Patau sono labio e palatoschisi, difetti del cuoio capelluto. In tutti i casi della sindrome è interessato il sistema nervoso centrale; sono frequenti l'aplasia e l'ipoplasia del verme cerebellare. Questa sindrome è caratterizzata dalle seguenti manifestazioni: microcefalia, trigonocefalia, fronte bassa e inclinata, naso largo con radice infossata, rime palpebrali strette, ipertelorismo, microftalmia (raramente anoftalmia), coloboma dell'iride, polidattilia delle mani e dei piedi, flessori posizione delle mani, piede a dondolo, criptorchidismo, ipospadia, ipoplasia del pene, sdoppiamento dell'utero e della vagina. Anomalie multiple degli organi interni, del sistema cardiovascolare (difetti del setto interventricolare e interatriale, difetti dei grandi vasi). I bambini affetti dalla sindrome di Patau muoiono nella maggior parte dei casi nei primi giorni e mesi di vita a causa di gravi malformazioni incompatibili con la vita. In casi isolati di aspettativa di vita fino a 2-3 anni, si osserva una profonda idiozia.
Sindrome di Edwards, trisomia del cromosoma 18. I bambini nascono con ipoplasia prenatale (il peso corporeo alla nascita non è superiore a 2200). Le manifestazioni fenotipiche della sindrome di Edwards sono caratteristiche e diverse. Questi includono dolicocefalia, microftalmia, padiglioni auricolari deformati in basso, palato alto, palatoschisi, microgenia, microstomia, ipertrofia del clitoride, ipospadia, criptorchidismo, anomalie degli arti (posizione dei flessori delle mani, alluce corto e largo, piede a dondolo, sindattilia cutanea del i piedi, piede torto). Tra i difetti degli organi interni si osservano anomalie del cuore (difetti del setto interatriale e interventricolare), della digestione, del sistema urinario, del sistema nervoso centrale (ipoplasia del cervelletto e del corpo calloso). In tutti i casi si osservano disturbi dello sviluppo cerebrale. Nella maggior parte dei casi, i bambini muoiono nel primo anno di vita. Rari casi di aspettativa di vita fino a 3-5 anni sono associati, di regola, a varianti a mosaico.
La sindrome più comune associata a delezioni autosomiche è sindrome del pianto del gatto monosomia del braccio corto del cromosoma 5 (5p-). Cariotipo 46,XX,del(5p), 46,XY,del(5p). Le caratteristiche principali sono uno specifico grido felino associato a cambiamenti nella laringe, che, di regola, scompare entro l'anno, microcefalia, viso a forma di luna, incisione anti-mongoloide degli occhi, epicanto, strabismo, ipertelorismo, piccolo mascella inferiore, padiglioni auricolari bassi, clinodattilia, sindattilia.
Altre sindromi da delezione relativamente comuni sono le sindromi di Wolf-Hirshhorn _ del(4p), del 13q di Orbeli, del 18(q) e del 18(p). Tra le sindromi della trisomia parziale, la più comune è la sindrome di Retore. Tutti sono caratterizzati da un forte ritardo nello sviluppo fisico e psicomotorio, un profondo ritardo mentale. In tutti i casi si osservano microcefalia di vario grado, dismorfismi facciali, anomalie del naso e degli occhi, padiglioni auricolari deformati e bassi. Tra gli organi interni, il cuore, i reni e gli organi del tratto gastrointestinale sono più spesso colpiti. Si possono osservare anomalie delle estremità. La prognosi per la vita è sfavorevole, la maggior parte dei pazienti muore nella prima infanzia. La combinazione di queste anomalie è la base per suggerire la presenza di uno squilibrio cromosomico. un'indicazione per il rinvio ad una consulenza genetica e ad un ulteriore esame citogenetico.
Malattie gonosomiali, a differenza di quelle autosomiche, raramente portano a gravi deviazioni dello stato di salute e dell'intelligenza del portatore di tali anomalie
MVPR n
Nessuna anomalia esterna multipla
L'intelligenza è normale, leggermente ridotta o qualitativamente alterata
Colpisce spesso i genitali esterni ed interni, l'altezza e il fisico
L’aspettativa di vita non è significativamente ridotta
Le malattie gonosomiali sono caratterizzate da un'ampia variabilità delle manifestazioni cliniche, dalla presenza di un fenotipo classico, che consente la "diagnostica del ritratto" e il sospetto immediato di anomalie del cariotipo, a manifestazioni relativamente lievi, quando il rinvio alla genetica viene effettuato solo dopo un lungo periodo trattamento infruttuoso dei problemi riproduttivi. Tuttavia, nella maggior parte dei casi, la malattia gonosomica può essere sospettata dai seguenti segni: a) deviazione della crescita. b) alterazioni del fisico, ritardo nello sviluppo sessuale, anomalie dei genitali esterni ed interni. c) negli adulti, segni di anomalie cromosomiche possono essere infertilità, irregolarità mestruali nelle donne, azoospermia negli uomini.
Sindrome di Klinefelter. Una variante dovuta alla disomia del cromosoma X. Cariotipo 47,XXY. Fenotipo maschile. La crescita è diversa. Tipo di corporatura eunucoide, crescita orizzontale dei peli pubici, ginecomastia. I genitali esterni sono formati secondo il tipo maschile, ma ipoplastici, la dimensione dei testicoli è nettamente ridotta ed è il microorchidismo ad essere considerato uno dei criteri clinici importanti nella diagnosi della sindrome. I pazienti con cariotipo 47,XXY presentano infertilità assoluta causata dalla ialinizzazione dei tubuli seminiferi e azoospermia. I pazienti con forme a mosaico possono essere fertili. La maggior parte dei pazienti è caratterizzata da un orientamento psicosessuale maschile. La funzione sessuale è preservata. L'intelligenza varia da praticamente invariata (nella maggior parte dei casi) al ritardo mentale. Circa il 25% dei casi della malattia vengono diagnosticati solo negli adulti, quando si fa riferimento alla genetica in relazione all'infertilità, una percentuale significativa di pazienti rimane non diagnosticata. Le forme polisomiche della sindrome di Klinefelter, quando nel cariotipo compaiono due o tre cromosomi X aggiuntivi, hanno un decorso più grave e sono accompagnate da ritardo mentale.
Sindrome di Shereshevskij-Turner.. Cariotipo 45,X o cromosoma X strutturalmente alterato. Tra i disturbi strutturali, le varianti citogenetiche più comuni sono: 46,X,i(X)(q10), 46,X,del(X), 46,X,r(X), forme a mosaico si osservano in quasi la metà dei casi i casi. fenotipo femminile. Bassa statura, sviluppo sessuale ritardato. Ipoplasia/aplasia dell'utero e delle ovaie. Nella maggior parte dei casi, amenorrea e infertilità. Anomalie esterne: ipertelorismo dei capezzoli e capezzoli introflessi, deformità in valgo delle articolazioni del gomito, pieghe pterigoidee del collo, alterazioni dell'arco digitale sui palmi e sui piedi, torace a scudo, bassa crescita dei peli sulla parte posteriore della testa, ecc. Nel 10-20% dei casi si osserva coartazione dell'aorta. Anomalie esterne si riscontrano solitamente nei casi di monosomia X completa, con anomalie strutturali sono meno pronunciate o del tutto assenti. L'intelligenza è normale. L'aspettativa di vita è leggermente ridotta. Nelle anomalie esterne la diagnosi può essere fatta già alla nascita, mentre i neonati spesso presentano edema linfatico delle mani e dei piedi. In assenza di anomalie esterne pronunciate, la causa del trattamento nelle ragazze in età prescolare può essere un pronunciato ritardo della crescita, in età avanzata - un ritardo nello sviluppo sessuale. Circa il 10% dei casi viene diagnosticato solo negli adulti, solitamente durante un esame per l'infertilità.
Sindrome XYY. Frequenza 1:1000. Cariotipo 47,XYY. Fenotipo maschile. Di norma, sono fertili, sebbene si trovino spesso tra i pazienti con infertilità. A causa dell'assenza di manifestazioni cliniche pronunciate, viene raramente diagnosticata.
Sindrome della trisomia X. Frequenza 1:1000. Cariotipo 47,XXX. fenotipo femminile. Crescita normale. Caratteristiche fisiche e facciali corrette. Vari disturbi mestruali, spesso amenorrea secondaria, insufficienza ovarica prematura. A volte c'è una leggera diminuzione dell'intelligenza. I pazienti, di regola, sono fertili, ma in età riproduttiva più avanzata è possibile l'infertilità, associata all'estinzione precoce della funzione ovarica. Raramente diagnosticato.
Tra i feti morti perinatalmente, la frequenza delle anomalie cromosomiche è del 6%.
Le forme più gravi di squilibrio cromosomico si riscontrano negli aborti precoci. Si tratta di poliploidie (25%), trisomie complete per autosomi (50%). Le trisomie per alcuni autosomi (1; 5; 6; 11; 19) sono estremamente rare anche negli embrioni e nei feti eliminati, il che indica il grande significato morfogenetico dei geni in questi autosomi. Queste anomalie interrompono lo sviluppo nel periodo preimpianto o interrompono la gametogenesi.
L'elevato significato morfogenetico degli autosomi è ancora più pronunciato nella monosomia autosomica completa. Questi ultimi si ritrovano raramente anche nel materiale degli aborti spontanei precoci a causa dell'effetto letale di un tale squilibrio.
Malformazioni congenite
Se un'anomalia cromosomica non ha un effetto letale nelle prime fasi dello sviluppo, le sue conseguenze si manifestano sotto forma di malformazioni congenite. Quasi tutte le anomalie cromosomiche (eccetto quelle bilanciate) portano a malformazioni congenite
sviluppo, le cui combinazioni sono note come forme nosologiche di malattie e sindromi cromosomiche (sindrome di Down, sindrome di Wolf-Hirshhorn, grido di gatto, ecc.).
Gli effetti causati dai disomogenei uniparentali possono essere trovati più dettagliatamente sul CD nell'articolo di S.A. Nazarenko "Malattie ereditarie determinate da disomi uniparentali e loro diagnostica molecolare".
Effetti delle anomalie cromosomiche nelle cellule somatiche
Il ruolo delle mutazioni cromosomiche e genomiche non si limita alla loro influenza sullo sviluppo di processi patologici nei primi periodi dell'ontogenesi (mancato concepimento, aborto spontaneo, natimortalità, malattia cromosomica). I loro effetti possono essere rintracciati per tutta la vita.
Le anomalie cromosomiche che si verificano nelle cellule somatiche nel periodo postnatale possono causare varie conseguenze: rimanere neutrali per la cellula, causare la morte cellulare, attivare la divisione cellulare, cambiare funzione. Le anomalie cromosomiche si verificano costantemente nelle cellule somatiche con una bassa frequenza (circa il 2%). Normalmente, tali cellule vengono eliminate dal sistema immunitario se si manifestano come estranee. Tuttavia, in alcuni casi (attivazione di oncogeni durante traslocazioni, delezioni), le anomalie cromosomiche causano una crescita maligna. Ad esempio, una traslocazione tra i cromosomi 9 e 22 causa la leucemia mieloide. L'irradiazione e gli agenti mutageni chimici inducono aberrazioni cromosomiche. Tali cellule muoiono e, insieme all'azione di altri fattori, contribuiscono allo sviluppo della malattia da radiazioni e dell'aplasia del midollo osseo. Esistono prove sperimentali dell'accumulo di cellule con aberrazioni cromosomiche durante l'invecchiamento.
Le cause di questo gruppo di malattie sono i cambiamenti nel numero degli autosomi (come la monosomia o la trisomia) o varie mutazioni cromosomiche degli autosomi. I disturbi più comuni sono i cromosomi 8, 9, 13 (sindrome di Patau), 18 (sindrome di Edwards), 21 (sindrome di Down).
Per diagnosticare la malattia, è necessario determinare:
Tipo di mutazione (genomica o cromosomica)
Il cromosoma coinvolto nel processo
Forma (intera o mosaico)
Tipo di malattia (sporadica o ereditaria)
Le mutazioni autosomiche causano una violazione dell'equilibrio genetico complessivo delle cellule, pertanto gli effetti patologici si manifestano in tutte le fasi dell'ontogenesi e in vari organi. Gli effetti principali delle mutazioni si manifestano in due modi: letalità e malformazioni congenite. L'effetto letale è uno dei principali fattori di morte fetale intrauterina. I bambini nati vivi presentano numerose disabilità dello sviluppo. Con varie anomalie cromosomiche si osservano manifestazioni cliniche simili: basso peso alla nascita, ritardo nello sviluppo fisico e mentale, alterata formazione del cranio facciale e cerebrale (deformazione dei padiglioni auricolari, microcefalia, ernie cerebrali),
malformazioni degli organi interni. Se alla nascita un bambino presenta diverse malformazioni, prima di tutto dovresti pensare alla presenza di una malattia cromosomica in lui.
Sindrome di Down. Avviene con una frequenza di 1:700-1:800 nascite, mentre non c'è dipendenza dal sesso del bambino. Il rischio di avere un figlio malato aumenta con l'età della madre e in misura minore dipende dall'età del padre.
Le varianti citogenetiche della malattia sono diverse: 95% - trisomia completa (mentre il "contributo materno" è dell'80%), forma di traslocazione (cromosoma 21 + cromosoma 15) - questa forma della malattia può essere ereditata dai genitori.
I bambini malati di razze diverse sono sorprendentemente simili tra loro. I pazienti hanno un aspetto caratteristico: una fessura mongoloide degli occhi, una faccia rotonda e appiattita, la parte posteriore del naso piatta, una lingua grande (solitamente sporgente) e orecchie deformate. I tratti dermatoglifici sono caratteristici: piega a “scimmia” sul palmo, due pieghe cutanee sul mignolo. Nei pazienti vengono rilevate malformazioni degli organi interni, più spesso del sistema cardiovascolare, degli organi digestivi e dei reni.
Nei pazienti lo sviluppo fisico e mentale è disturbato: la crescita dei pazienti adulti è di 20 cm inferiore alla media, ritardo mentale (oligofrenia) di vario grado. I pazienti sono affettuosi, obbedienti, possono padroneggiare semplici abilità pratiche. Tuttavia, una diminuzione del tono muscolare, combinata con la scioltezza delle articolazioni, non consente movimenti fini e coordinati. L’aspettativa di vita media è di 30-35 anni.
Sindrome del "grido del gatto" per la perdita (delezione) di un tratto del braccio corto del cromosoma 5 (cariotipo: 46, 5 p-). I bambini hanno un grido insolito, che ricorda il miagolio di un gatto esigente. Il quadro clinico varia considerevolmente nei diversi pazienti. Il pianto caratteristico è dovuto a un cambiamento della laringe (restringimento, morbidezza della cartilagine, diminuzione dell'epiglottide). Quasi tutti i pazienti presentano cambiamenti nella struttura del cervello e del cranio facciale: faccia a forma di luna, microcefalia. I padiglioni auricolari sono deformati e localizzati in basso. Sono presenti difetti del cuore e del sistema muscolo-scheletrico: sindattilia, piede torto. La maggior parte dei pazienti muore prima dell'età di 1 anno.
Nel caso dell'eteroploidia, la monosomia è particolarmente grave. La monosomia per gli autosomi termina mortalmente nei primi giorni dello sviluppo embrionale o porta alla morte dell'embrione nelle fasi successive (aborto spontaneo). Le trisomie complete sono descritte nell'uomo per un gran numero di cromosomi: 8, 9, 13, 14, 18, 21, X, Y. Le sindromi più studiate basate su disturbi del sistema autosomico (mutazioni genomiche, mutazioni cromosomiche) sono le trisomie 21 , 13 , 18, Forma di traslocazione di Down, sindrome del "grido di gatto", trisomia XXY, XXX, XYY e monosomia XO nel sistema dei cromosomi sessuali.
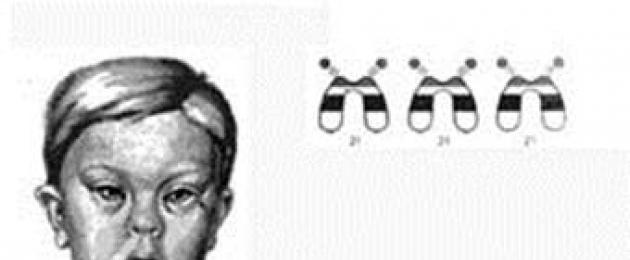
malattia di Down
(trisomia 21; 47,XX(XY)+21)
La diagnosi della malattia di Down già in un neonato non causa difficoltà (Fig. 5). Nella malattia di Down si verificano da 9 a 29 anomalie somatiche. Più spesso con questa sindrome ci sono:
Cranio brachicefalo con occipite appiattito e faccia appiattita, epicanto;
Macchie di Brushfield (macchie luminose sull'iride);
· Piccoli padiglioni auricolari sottosviluppati;
Lingua "piegata" allargata;
· Pennelli larghi con dita corte e quinto dito ricurvo accorciato (clinodattilia);
· Solco trasversale su uno o entrambi i palmi ("piega della scimmia");
· Spazi estesi tra il primo e il secondo dito del piede.
 |
| Fig.5. Sintomi della trisomia 21 |
Il difetto intellettuale dei pazienti si approfondisce con l'età. È noto che circa il 60% dei bambini affetti dalla malattia di Down presenta varie forme di patologie oculari e il 70% soffre di perdita dell'udito.
Negli ultimi anni molta attenzione è stata rivolta allo studio della patogenesi della sindrome di Down. Oggi è stata proposta un'ipotesi genetica combinata per la sindrome di Down e il morbo di Alzheimer. Lo stato di questi pazienti rivela invecchiamento precoce, predominanza di disturbi vascolari degenerativi, diabete mellito, cataratta, lipofuscinosi, amiloidosi, danno selettivo ai neuroni colinergici nei gangli della base, tendenza a neoplasie maligne, disturbi uditivi specifici e altri segni, e la maggior parte soprattutto, disabilità intellettive caratteristiche. ͵ che ricordano quelle della malattia di Alzheimer senile.
L'uso di metodi di ricerca citogenetica ha dimostrato che circa l'80% di tutti i casi di trisomia 21 semplice sono di origine materna e circa il 20% di origine paterna. Allo stesso tempo, solo il 20% di tutti i casi di sindrome di Down "materna" sono dovuti alla mancata disgiunzione dei cromosomi della 21a coppia nella seconda divisione della meiosi, e il resto è dovuto a errori nella prima divisione della meiosi.
Malattia di Down della forma di traslocazione
Forme di traslocazione della sindrome di Down si osservano nel 3-4% dei casi. Il numero di cromosomi in questa variante della malattia è normale - 46, poiché il cromosoma aggiuntivo 21 viene traslocato negli autosomi 13, 14, 15 e 22 (Fig. 6)
Nel caso dell'eteroploidia, la monosomia è particolarmente grave. La monosomia per gli autosomi termina mortalmente nei primi giorni dello sviluppo embrionale o porta alla morte dell'embrione nelle fasi successive (aborto spontaneo). Le trisomie complete sono descritte nell'uomo per un gran numero di cromosomi: 8, 9, 13, 14, 18, 21, X, Y. Le sindromi più studiate basate su disturbi del sistema autosomico (mutazioni genomiche, mutazioni cromosomiche) sono le trisomie 21 , 13 , 18, Forma di traslocazione di Down, sindrome del "grido di gatto", trisomia XXY, XXX, XYY e monosomia XO nel sistema dei cromosomi sessuali.
malattia di Down
(trisomia 21; 47,XX(XY)+21)
La diagnosi della malattia di Down già in un neonato non causa difficoltà (Fig. 5). Nella malattia di Down si verificano da 9 a 29 anomalie somatiche. Più spesso con questa sindrome ci sono:
Cranio brachicefalo con occipite appiattito e faccia appiattita, epicanto;
Macchie di Brushfield (macchie luminose sull'iride);
· Piccoli padiglioni auricolari sottosviluppati;
Lingua "piegata" allargata;
· Pennelli larghi con dita corte e quinto dito ricurvo accorciato (clinodattilia);
· Solco trasversale su uno o entrambi i palmi ("piega della scimmia");
· Spazi estesi tra il primo e il secondo dito del piede.
| Fig.5. Sintomi della trisomia 21 |
Il difetto intellettuale dei pazienti si approfondisce con l'età. È noto che circa il 60% dei bambini affetti dalla malattia di Down presenta varie forme di patologie oculari e il 70% soffre di perdita dell'udito.
Negli ultimi anni molta attenzione è stata rivolta allo studio della patogenesi della sindrome di Down. È stata ora proposta un'ipotesi genetica combinata per la sindrome di Down e il morbo di Alzheimer. Lo stato di questi pazienti rivela invecchiamento precoce, predominanza di disturbi vascolari degenerativi, diabete mellito, cataratta, lipofuscinosi, amiloidosi, danno selettivo ai neuroni colinergici nei gangli della base, tendenza a neoplasie maligne, disturbi uditivi specifici e altri segni, e la maggior parte soprattutto, disturbi intellettivi caratteristici simili a quelli della malattia di Alzheimer senile.
L'uso di metodi di ricerca citogenetica ha dimostrato che circa l'80% di tutti i casi di trisomia 21 semplice sono di origine materna e circa il 20% di origine paterna. Allo stesso tempo, solo il 20% di tutti i casi di sindrome di Down "materna" sono dovuti alla mancata disgiunzione dei cromosomi della 21a coppia nella seconda divisione della meiosi, e il resto è dovuto a errori nella prima divisione della meiosi.
Malattia di Down della forma di traslocazione
Forme di traslocazione della sindrome di Down si osservano nel 3-4% dei casi. Il numero di cromosomi in questa variante della malattia è normale - 46, poiché il cromosoma aggiuntivo 21 viene traslocato negli autosomi 13, 14, 15 e 22 (Fig. 6)
Nella variante di traslocazione della sindrome di Down, uno dei genitori fenotipicamente sani può essere portatore di una traslocazione equilibrata. Il cariotipo di questi genitori ha 45 cromosomi. Un cromosoma è costituito, per così dire, da due parti e contiene il materiale genetico del cromosoma mancante (Fig. 8), quindi, con un numero totale di cromosomi pari a 45, non vi è alcuna perdita di materiale genetico e il riarrangiamento è equilibrato. In circa 1/3 dei casi, la variante di traslocazione della sindrome di Down è ereditaria. L'identificazione di una traslocazione equilibrata in uno dei genitori determina la necessità della diagnosi prenatale.
sindrome di Edwards
(trisomia 18; 47,XX(XY)+18)
Durante l'esame cariologico dei pazienti viene rilevato un cromosoma in più del gruppo E (cromosoma 18) (Fig. 7).
 |
| Riso. 7. Sintomi della trisomia 18 |
Le manifestazioni fenotipiche della sindrome di Edwards sono piuttosto caratteristiche:
· Cranio dolicocefalo, compresso lateralmente, con fronte bassa e occipite largo e sporgente;
Le rime palpebrali sono strette; epico;
La mascella inferiore è piccola, smussata all'indietro (microretrognazia);
La bocca è piccola, di forma triangolare con labbro superiore corto;
Il collo è corto, con una piega pterigoidea.
Anomalie del sistema muscolo-scheletrico:
- Le mani e le dita sono corte, il quinto dito è curvo, le dita sono serrate a pugno, il secondo e il quinto dito si trovano in alto e coprono il secondo e il quarto dito premuti sul palmo;
- Il primo dito è corto e largo, sindattilia del secondo e terzo dito;
- La forma del piede a forma di "sedia a dondolo".
Quasi il 95% dei pazienti presenta malformazioni del cuore, dei grandi vasi, del sistema genito-urinario e anomalie dell'apparato digerente. La prognosi per la vita è sfavorevole.
sindrome di Patau
(trisomia 13; 47,XX(XY)+13)
L'analisi cariologica delle cellule somatiche dei pazienti ha rivelato un cromosoma in più del gruppo D (cromosoma 13) (Fig. 8).
Il quadro clinico è tipico:
· Cranio microcefalico con fronte bassa e inclinata e regioni temporali depresse;
Le rime palpebrali sono strette, disposte orizzontalmente, la distanza tra loro è ridotta (ipotelorismo), si incontra quasi sempre patologia oculare;
I padiglioni auricolari sono situati bassi, piccoli lobi sono premuti sulla testa, riccioli di forma irregolare;
· Cranio con recessi nella regione parieto-occipitale, la distanza tra i tubercoli parietali è aumentata;
Il segno dimostrativo della sindrome di Patau è il "labbro leporino" e la "palatoschisi". Le schisi possono essere bilaterali o unilaterali. Quasi sempre, la spaccatura del labbro superiore è accompagnata da una palatoschisi.
 |
| Riso. 8. Sintomi della trisomia 13 |
Anomalie del sistema muscolo-scheletrico:
Polidattilia sugli arti superiori e inferiori;
Il secondo e il quarto dito vengono piegati, portati al palmo e bloccati dal primo e dal quinto dito;
Vengono rivelati difetti nello sviluppo di quasi tutti i sistemi e organi;
· Il cervello spesso non è diviso in emisferi, ipoplasia dei lobi frontali, cervelletto.
Nel 50% dei pazienti si riscontrano malformazioni delle vie urinarie: rene cistico, idronefrosi, displasia renale, nel 50% delle ragazze si riscontra uno sdoppiamento della vagina e un utero bicorne con ipoplasia ovarica. La prognosi per la vita è sfavorevole.
Sindrome del "grido del gatto"
(sindrome 5r–)
La più comune di tutte le sindromi da delezione autosomica è la sindrome da delezione del braccio corto del cromosoma 5. Nei pazienti con analisi cariologica si riscontra un accorciamento del braccio corto di uno dei cromosomi del gruppo B (Fig. 9)
Le caratteristiche fenotipiche della sindrome sono:
· Microcefalia;
Viso tondo "a luna" nei primi anni di vita e viso stretto in età avanzata;
Incisione antimongoloide degli occhi, epicanto, strabismo, cataratta, focolai di pigmentazione retinica, atrofia dei nervi ottici;
Ponte del naso piatto, palato alto;
Le orecchie sono deformate;
Sindattilia delle dita dei piedi, piede torto, ipotensione muscolare;
Un sintomo peculiare è il pianto alla nascita, che ricorda il pianto di un gatto. È presente nei bambini del primo anno di vita. È causato da una violazione dell'attività del sistema nervoso centrale e da cambiamenti nella laringe (riduzione dell'epiglottide, restringimento della laringe, gonfiore della mucosa).
La prognosi per la vita dipende dalla gravità dei sintomi. Molti pazienti sopravvivono fino all’adolescenza. Il ritardo mentale è sempre profondo. La diagnosi finale viene stabilita come risultato dello studio del cariotipo.
- In contatto con 0
- Google+ 0
- OK 0
- Facebook 0






