RCHR (Centro Repubblicano per lo Sviluppo Sanitario del Ministero della Salute della Repubblica del Kazakistan)
Versione: Protocolli clinici del Ministero della Salute della Repubblica del Kazakistan - 2015
Sindromi mielodisplastiche (D46)
Oncoematologia
informazioni generali
Breve descrizione
Consigliato
Consigli degli esperti
RSE presso il RVC “Centro Repubblicano”
sviluppo sanitario"
ministero della Salute
e sviluppo sociale
Repubblica del Kazakistan
del 9 luglio 2015
Protocollo n. 6
Definizione: La sindrome mielodisplastica è un gruppo di malattie clonali eterogenee caratterizzate dalla presenza di citopenia nel sangue periferico, displasia del midollo osseo e rischio di trasformazione in leucemia acuta.
|
Nome del protocollo: Sindrome mielodisplastica negli adulti. |
||||||||||||
|
Codice protocollo: Codice ICD-10: |
||||||||||||
|
Data di sviluppo del protocollo: 2015 Abbreviazioni utilizzate nel protocollo: |
||||||||||||
|
Ara-C - citarabina BUN-(Azoto ureico nel sangue) azoto ureico DNR - daunorubicina FAB - classificazione - sistema di classificazione franco-americano-britannico FISH - ibridazione fluorescente in situ IgG - immunoglobulina G HLA - sistema antigene leucocitario umano Ida - idarubicina IPSS - Sistema internazionale di punteggio prognostico NCCN- Rete nazionale completa contro il cancro PICC - catetere centrale inserito perifericamente Pro-BNP ( proormone peptide natriuretico cerebrale) - ormone natriuretico cerebrale Sistema di punteggio prognostico WPSS-OMS. |
||||||||||||
|
Categoria paziente: pazienti adulti Scala del livello di evidenza.
|
Classificazione
Classificazione clinica:
Caratteristiche dei diversi tipi di MDS secondo la classificazione dell'OMS, 2001.
| Tipo MDS | Cambiamenti nel sangue | Cambiamenti nel midollo osseo |
| Anemia refrattaria | Anemia, meno dell'1% di esplosioni | Displasia della linea eritroide, meno del 5% di blasti |
| Anemia refrattaria con sideroblasti ad anello (RAS) | Lo stesso di RA | Come l'AR, ≥ 15% di sideroblasti ad anello |
| Citopenia refrattaria con displasia multilineare (RCMD) | Citopenia per 2-3 germogli, meno dell'1% dei blasti | Displasia, più del 10% di cellule di 2 o 3 linee, meno del 5% di blasti, meno del 15% di sideroblasti ad anello |
| Citopenia refrattaria con displasia multilineare e sideroblasti ad anello (RCMD-CS) | Lo stesso dell'RCMD | Come RCMD, ≥ 15% di sideroblasti ad anello |
| Anemia refrattaria con eccesso di blasti, tipo I (RAEB-1) | Citopenia, meno del 5% di esplosioni | 5-9% esplosioni |
| Anemia refrattaria con eccesso di blasti, tipo II (RAEB-2) | Citopenia, 5-19% di esplosioni | 10-19% esplosioni |
| Sindrome 5q- | Anemia, conta piastrinica normale o elevata | Numero normale o aumentato di megacariociti con nuclei iposegmentati; delezione isolata 5q31 |
| MDS non classificati (MDS-N) | Citopenia | Displasia unilineare nelle linee neutrofile o megacariocitiche, blasti inferiori al 5%, assenza di bastoncini di Auer |
Indicatori prognostici nei pazienti con MDS a seconda del grado di rischio secondo la scala IPSS.
| Somma dei punti | Rischio IPSS | % di pazienti | ||
| 0 | Corto | 9,4 | 5,7 | 31 % |
| 0,5-1,0 | Intermedio-1 | 3,3 | 3,5 | 39 % |
| 1,5-2,0 | Intermedio-2 | 1,1 | 1,2 | 22 % |
| ≥ 2,5 | Alto | 0,2 | 0,4 | 8 % |
Indicatori prognostici nei pazienti con MDS a seconda del grado di rischio secondo la scala IPSS-R.
| Somma dei punti | Rischio IPSS | Tempo alla conversione in LMA nel 25% dei pazienti (anni) | Sopravvivenza globale mediana (anni) | % di pazienti |
| ≤1.5 | Molto basso | Non raggiunto | 8,8 | 13 % |
| >1,5≤3,0 | Corto | 10,8 | 5,3 | 38 % |
| >3,0≤4,5 | Intermedio | 3,2 | 3 | 20 % |
| >4,5≤6,0 | Alto | 1,4 | 1,6 | 13 % |
| >6,0 | Molto alto | 0,7 | 0,8 | 10 |
Determinazione del gruppo di rischio utilizzando la scala WPSS.
| Punti | 0 | 1 | 2 | 3 |
| Tipo di MDS secondo la classificazione dell'OMS | RA, RAX, 5q- | RCMD, RCMD-KS | RAIB1 | RAEB2 |
| Cariotipo | Bene | Media | Cattivo | - |
| Necessità di trasfusioni di sangue | NO | Regolare | - | - |
Cariotipo:
· Buono: normale, -Y, del 5q, del 20q;
· Scarso: più di 3 anomalie o anomalie del cromosoma 7;
· Medio: tutti gli altri.
Trasfusioni di sangue regolari: trasfusione di almeno 1 dose di globuli rossi ogni 8 settimane per 4 mesi.
Diagnostica
Elenco delle misure diagnostiche di base e aggiuntive:
Esami diagnostici di base (obbligatori) eseguiti in regime ambulatoriale:
· esame del sangue generale (contaggio della leucemia, dei reticolociti, delle piastrine in uno striscio);
· mielogramma;
· Studio FISH e studio di genetica molecolare (-7/7q-; EGR1(5q); +20q; -Y).
Ulteriori accertamenti diagnostici eseguiti in regime ambulatoriale:
· analisi del sangue biochimiche (proteine totali, albumina, bilirubina totale, bilirubina diretta, creatinina, urea, ALT, ACaT, glucosio, LDH, proteina C-reattiva, fosfatasi alcalina), studi sul metabolismo del ferro, vitamina B-12 e livelli di acido folico ;
· coagulogramma;
· gruppo sanguigno e fattore Rh;
· ELISA per i marcatori dell'HIV;
· ELISA per marcatori dei virus del gruppo dell'herpes;
· analisi generale delle urine;
· esame istologico del campione bioptico trapanato della cresta iliaca;
· ECG;
L'elenco minimo degli esami da effettuare in caso di ricovero programmato:
· esame del sangue biochimico (proteine totali, albumina, bilirubina totale, bilirubina diretta, creatinina, urea, ALT, ACaT, glucosio, LDH, proteina C-reattiva);
· mielogramma;
· Ecografia degli organi addominali e della milza;
· Ultrasuoni degli organi pelvici - per le donne.
Esami diagnostici di base (obbligatori) effettuati a livello ospedaliero:
· esame del sangue generale (calcolo della leucemia, piastrine in uno striscio);
· esame del sangue biochimico (proteine, albumina, ALT, ACaT, bilirubina, fosfatasi alcalina, GGTP, creatinina, urea, acido urico, elettroliti, LDH, glucosio, proteina C reattiva, immunoglobuline G, A, M, ferritina, sideremia) ;
· coagulogramma;
· determinazione dell'antitrombina III nel sangue plasmatico;
· determinazione quantitativa del livello di D - dimeri nel plasma sanguigno;
· mielogramma;
· studio citochimico dei blasti (MPO, glicogeno, alfa
NE, Sudan nero);
· test immunologici (immunofenotipizzazione per escludere emoglobinuria parossistica notturna);
· “pannello per leucemia acuta” di immunofenotipizzazione mediante citometria a flusso;
· studio citogenetico standard;
· Ricerca sulla FISH e ricerca genetica molecolare;
· gruppo sanguigno e fattore Rh;
· ELISA per marcatori di epatite virale;
· ELISA per i marcatori dell'HIV;
· analisi generale delle urine;
· Test di Rehberg;
· Radiografia degli organi del torace.
Ulteriori esami diagnostici effettuati a livello ospedaliero:
· pro-BNP (peptide natriuretico atriale) nel siero del sangue;
· PCR per le infezioni virali (epatite virale, citomegalovirus, virus dell'herpes simplex, virus Epstein-Barr, virus Varicella/Zoster);
· HLA - tipizzazione;
· esame batteriologico del materiale biologico;
· esame citologico del materiale biologico;
· immunogramma;
· esame istologico del materiale bioptico (linfonodo, cresta iliaca);
· esame immunoistochimico del campione bioptico (cresta iliaca);
· esame del liquido cerebrospinale;
· ecocardiografia;
· Ultrasuoni degli organi addominali (fegato, milza, pancreas, cistifellea, linfonodi, reni), nelle donne - bacino;
Radiografia dei seni paranasali;
· radiografia delle ossa e delle articolazioni;
· TAC del segmento toracico, del segmento addominale, della testa, del bacino;
· RM del segmento toracico, del segmento addominale, della testa, del bacino;
· FGDS;
· Ecografia Doppler dei vasi;
· broncoscopia;
· colonscopia;
· Monitoraggio della pressione arteriosa 24 ore su 24;
· Monitoraggio ECG 24 ore su 24;
· spirografia.
Misure diagnostiche effettuate nella fase di assistenza medica di emergenza:
· raccolta dei reclami e dell'anamnesi;
· esame fisico (determinazione del RR, frequenza cardiaca, valutazione della pelle, determinazione delle dimensioni del fegato e della milza).
Criteri diagnostici per la diagnosi:
Reclami su:
- debolezza;
- sudorazione;
- stanchezza;
- febbre bassa;
- agghiacciante;
- perdita di peso;
- eruzioni emorragiche sotto forma di petecchie ed ecchimosi sulla pelle;
- epistassi;
-menorragia;
- aumento del sanguinamento.
Anamnesi:
dovresti prestare attenzione a:
- debolezza a lungo termine;
- affaticamento rapido;
- frequenti malattie infettive;
- aumento del sanguinamento;
- la comparsa di eruzioni cutanee emorragiche sulla pelle e sulle mucose.
Esame fisico[
5
]
:
- pallore della pelle;
- eruzioni emorragiche - petecchie, ecchimosi;
- fiato corto;
- tachicardia;
- milza ingrossata.
Criteri diagnostici per la diagnosi:
I criteri diagnostici minimi per le MDS includono condizioni diagnostiche obbligatorie (citato da NCCN, 2.2015):
· citopenia stabile per almeno 6 mesi (ad eccezione dei casi in cui la citopenia è accompagnata da un cariotipo specifico o da displasia di due linee ematopoietiche - in questi casi la durata della citopenia stabile dovrebbe essere di almeno 2 mesi);
· esclusione di altre malattie che possono causare lo sviluppo di displasia e/o citopenia.
Oltre a queste due condizioni diagnostiche, per fare una diagnosi di MDS deve essere soddisfatto almeno uno dei tre criteri principali:
· displasia (≥ 10% delle cellule di una o più delle tre principali linee ematopoiesi nel midollo osseo);
· il contenuto di blasti nel midollo osseo è del 5-19%;
· cariotipo specifico, ad esempio, delezione (5q-), delezione (20q-), +8 o −7/delezione (7q-).
Ricerca di laboratorio:
Studio morfologico: i segni morfologici di displasia di elementi di vari germi ematopoietici sono il fattore determinante nel riconoscere la MDS. Le manifestazioni displastiche più tipiche per la diagnosi di MDS sono: :
· La presenza di neutrofili ipo- o ipergranulari e la conseguente carenza di attività perossidasica in tutti gli stadi di maturazione delle cellule della mielopoiesi;
· pelgeroidità delle cellule della serie neutrofila ed eosinofila;
· presenza di più del 15% di sideroblasti ad anello;
· PAS - materiale positivo nelle cellule megaloblastoidi;
· micromegacariociti con un nucleo picnotico o megacariociti giganti con molti nuclei rotondi separati.
Immunofenotipizzazione: Gli studi immunofenotipici non hanno rivelato alcun marcatore specifico di MDS. Questo studio nella MDS viene effettuato per escludere l'emoglobinuria parossistica notturna (nella MDS, nel 10-15% dei casi, il clone PNH è positivo), e l'IFT consente anche un calcolo più accurato del numero di blasti.
Esame istologico: caratterizzato da una violazione della topografia del midollo osseo di tutti i germi ematopoietici. Istologicamente si distinguono due opzioni:
MDS con aumentata cellularità del midollo osseo/BM ipercellulare: Si osserva un leggero aumento dell'eritropoiesi con aumento del numero di cellule immature della serie eritroblastica, principalmente proeritroblasti. Le cellule immature della serie eritroblastica formano isole contenenti almeno 10 cellule. Questi piccoli focolai di eritropoiesi sono combinati con cellule granulocitiche distribuite in modo relativamente uniforme nelle sezioni. Il numero di cellule megaloblastoidi aumenta.
MDS con ridotta cellularità del midollo osseo/BM ipocellulare: Nella linea dei megacariociti sono presenti accumuli di micromegacariociti, una violazione dell'orientamento sinusoidale e localizzazione paratrabecolare dei megacariociti displastici e picnotici. Una caratteristica della linea mieloide è la localizzazione patologica delle cellule immature - ALIP (localizzazione anormale dei precursori immaturi). Da parte dell'eritropoiesi si individuano aree con blocco maturativo, localizzate sia in sede intra che paratrabecolare
Studio citogenetico standard: cariotipo specifico, ad esempio, delezione (5q-), delezione (20q-), +8 o −7/delezione (7q-).
Ricerca genetica molecolare: per la ricerca sulle MDS con il metodo FISH - (-7/7q-; EGR1(5q); +20q; -Y) sono tipici.
Studi strumentali:
Ultrasuoni degli organi addominali: aumento delle dimensioni della milza.
TAC del segmento toracico: alterazioni infiltrative nel tessuto polmonare.
ECG: Difficoltà di conduzione degli impulsi nel muscolo cardiaco.
EcoCG: segni di insufficienza cardiaca (HF)<60%), снижение сократимости, диастолическая дисфункция, легочная гипертензия, пороки и регургитации клапанов.
Indicazioni per la consultazione con specialisti:
· medico per la diagnostica e la terapia endovascolare radiografica - installazione di un catetere venoso centrale da un accesso periferico (PICC);
· epatologo – per la diagnosi e il trattamento dell'epatite virale;
· ginecologo - gravidanza, metrorragia, menorragia, consultazione quando si prescrivono contraccettivi orali combinati;
Dermatovenerologo - sindrome della pelle
· specialista in malattie infettive - sospetto di infezioni virali;
· cardiologo - ipertensione non controllata, insufficienza cardiaca cronica, disturbi del ritmo cardiaco e della conduzione;
· neurologo accidente cerebrovascolare acuto, meningite, encefalite, neuroleucemia;
· neurochirurgo - accidente cerebrovascolare acuto, sindrome da lussazione;
· nefrologo (efferentologo) - insufficienza renale;
· oncologo - sospetto di tumori solidi;
· otorinolaringoiatra – per la diagnosi e la cura delle malattie infiammatorie dei seni paranasali e dell'orecchio medio;
· oculista - disturbi della vista, malattie infiammatorie dell'occhio e delle appendici;
· proctologo - ragade anale, paraproctite;
· psichiatra - psicosi;
· psicologo – depressione, anoressia, ecc.;
· rianimatore - trattamento della sepsi grave, dello shock settico, della sindrome da danno polmonare acuto con sindrome da differenziazione e condizioni terminali, installazione di cateteri venosi centrali.
· reumatologo - sindrome di Sweet;
· chirurgo toracico - pleurite essudativa, pneumotorace, zigomicosi polmonare;
· trasfusiologo – per la scelta del mezzo trasfusionale in caso di positività al test antiglobulina indiretto, inefficacia delle trasfusioni, perdita ematica acuta e massiva;
· urologo - malattie infettive e infiammatorie dell'apparato urinario;
· tisiatra - sospetto di tubercolosi;
· chirurgo - complicanze chirurgiche (infettive, emorragiche);
· chirurgo maxillo-facciale - malattie infettive e infiammatorie del sistema dentofacciale.
Diagnosi differenziale
Diagnosi differenziale
La diagnosi differenziale della sindrome mielodisplastica si effettua con:
· Anemia megaloblastica(malattie caratterizzate da cambiamenti nella morfologia delle cellule del midollo osseo dovute alla ridotta sintesi del DNA. Oltre il 90% - anemia da carenza di B-12 e folati). Dopo l'inizio della terapia con vitamina B-12 o acido folico, un esame del sangue rivela una crisi reticolare nei giorni 5-7 e un aumento della conta dei globuli rossi, che non è tipico dei pazienti con anemia refrattaria. I cambiamenti nel cariotipo delle cellule del midollo osseo non si verificano nell'anemia megaloblastica.
· Anemia aplastica può essere congenito, acquisito e idiopatico. Anemia aplastica congenita - L'anemia di Fanconi si associa ad altre anomalie genetiche (pigmentazione cutanea, ipoplasia renale, microcefalia), acquisita è associata all'azione di agenti chimici e fisici, infezioni e disordini metabolici. L'anemia aplastica non è caratterizzata da cambiamenti nel cariotipo e nel midollo osseo ipercellulare.
· Anemia in cronica l'epatite è caratterizzata dall'identificazione di marcatori di infezioni virali, epatosplenomegalia e quadro clinico dell'epatite cronica. epatite, alterazioni dei parametri biochimici del sangue (metabolismo della bilirubina, funzionalità epatica).
Cure all'estero
Ottieni cure in Corea, Israele, Germania, Stati Uniti
Ottieni consigli sul turismo medico
Trattamento
Obiettivi del trattamento:
· ottenere una risposta completa o parziale (criteri di risposta vedere clausola 15);
· riduzione della dipendenza dalle trasfusioni e regressione del sovraccarico di ferro, nei casi in cui non sia possibile ottenere una risposta (per i criteri di risposta vedere paragrafo 15).
Tattiche di trattamento:
Trattamento non farmacologico:
Modalità: sicurezza generale.
Dieta: tabella n. 15. Si sconsiglia ai pazienti neutropenici di seguire una dieta specifica ( livello di evidenza B).
Trattamento farmacologico:

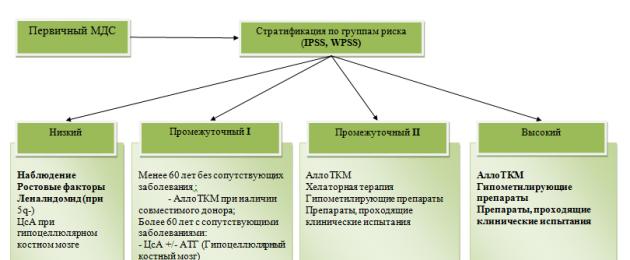
Secondo le raccomandazioni del NCCN (2.2015), la scelta del regime terapeutico si basa sulla categoria prognostica del paziente (IPSS, IPSS-R, WPSS). In base alla stratificazione dei pazienti per gruppi di rischio si distinguono due principali gruppi terapeutici:
· Gruppo terapeutico “a basso rischio”.:
IPSS: Basso, Intermedio -1;
IPSS-R: Molto basso, Basso, Intermedio;
WPSS: Basso, Molto basso, Intermedio.
· Gruppo terapeutico “ad alto rischio”:
IPSS: Intermedio-2, Alto;
IPSS-R: Intermedio, Alto, Molto Alto;
WPSS: Alto, Molto alto.
Terapia immunosoppressiva per le MDS (livello di evidenza B):
Le MDS possono svilupparsi contemporaneamente ad alcuni disturbi del sistema immunitario. I pazienti con MDS spesso presentano anomalie del sistema immunitario: autoanticorpi, disglobulinemia, cloni di cellule T autoreattive, squilibrio tra diverse popolazioni di cellule T, anomalie delle cellule NK, aumento della popolazione regolatoria T (CD4+25+; FoxP3+), aumento della produzione di IL-17 da parte delle cellule T. (citochine proinfiammatorie e proapoptotiche). In alcuni pazienti, le cellule NK e altri linfociti appartengono alla linea MDS.
La disregolazione immunitaria può precedere o predisporre allo sviluppo di malattie ematologiche clonali. Le reazioni immunitarie dirette contro le cellule staminali del midollo osseo possono accompagnare o essere alla base della MDS. I linfociti citotossici attivati e la funzionalità ridotta delle cellule T possono svolgere un ruolo nello sviluppo della MDS. I linfociti citotossici possono causare alterazioni aplastiche o displastiche nel midollo osseo, possono innescare l’apoptosi e possono causare rotture cromosomiche.
Nella variante ipoplastica (ipoproliferativa) della MDS viene utilizzata la terapia immunosoppressiva, per analogia con l'anemia aplastica. Le principali indicazioni all'uso dell'IST sono la conferma dell'ipoplasia emopoietica mediante esame istologico del midollo osseo, rischio basso o intermedio I secondo IPSS.
I principali farmaci IST per le MDS:
Ciclosporina;
Immunoglobulina antitimocitaria.
Terapia con ciclosporina:
1) 3-5 mg/kg/die in 2 dosi frazionate per os;
2) La concentrazione ottimale di ciclosporina nel sangue è fino a 300 ng/ml;
3) Effetto in pazienti con ipoplasia del midollo osseo e accumuli policlonali di elementi linfoidi nel midollo osseo, con cariotipo normale: 53-83%.
Immunoglobulina antitimocitaria.
Sono gammaglobuline purificate, principalmente IgG monomeriche, dal siero di cavalli, conigli e capre immunizzati rispettivamente con timociti o linfociti umani.
L'immunoglobulina antitimocitaria viene utilizzata sotto forma di infusione endovenosa, solitamente alla dose di 10-20-40 mg/kg di peso corporeo in 500-1000 ml di soluzione isotonica di cloruro di sodio per 3-18 ore per 5 giorni. Se l'attività delle cellule immunocompetenti del sangue e del midollo osseo è bassa, l'ATG può essere utilizzato in dosi di 5-10 mg/kg.
MDS con delezione 5q.
5q- è stato descritto da Vanden Berghe et al. nel 1974. Le caratteristiche cliniche della sindrome 5q descritte nel primo rapporto includevano macrocitosi, anemia, conta piastrinica normale o aumentata e megacariociti ipolobulari nel midollo osseo.
Lenalidomide.
Uso di lenalidomide nel trattamento di pazienti con MDS 5q- (Livello di evidenza B). La lenalidomide ha proprietà sia immunomodulatorie che antiangiogeniche. La dose iniziale raccomandata è di 25 mg una volta al giorno nei giorni 1-21 di cicli ripetuti di 28 giorni. Nel 67% dei pazienti affetti da MDS è stata riscontrata una diminuzione della dipendenza dalle trasfusioni di sangue.
Decitabina (livello di evidenza A). Tra gli inibitori dell'ipermetilazione, la decitabina è preferita per i pazienti anziani con rischio basso e intermedio 1 (scala IPSS). Gli studi hanno dimostrato che il farmaco aumenta il tempo prima della trasformazione della MDS in AML, riduce la dipendenza dalle trasfusioni, tuttavia, non influisce sulla sopravvivenza globale. È stata notata una tollerabilità abbastanza buona del farmaco.
Regimi di dosaggio della decitabina (DACO-022).
*Uguale randomizzazione fino a 45 pazienti. Ciclo ogni 4-6 settimane.
Inoltre, l’indicazione per la prescrizione di farmaci ipometilanti nel gruppo “a basso rischio” è la mancanza di risposta alle IST.
Inibitori dell'ipermetilazione.
Secondo gli studi, l'azacitidina è preferibile tra gli inibitori dell'ipermetilazione per i pazienti ad alto rischio (secondo la scala IPSS) e per i pazienti di età superiore a 75 anni, poiché l'uso del farmaco in questo gruppo non solo ha un effetto positivo sulla sopravvivenza globale (fino a a 24 mesi), ma aumenta i tempi di trasformazione di MDS in AML (fino a 21 mesi).
Azacitidina (livello di evidenza A)è destinato al trattamento di pazienti affetti da vari tipi di MDS ad alto rischio con eccesso di blasti. Secondo gli studi, nei gruppi ad alto rischio, non solo aumenta il tempo di trasformazione, ma anche la sopravvivenza complessiva (fino a 6 mesi).
Per il primo ciclo di terapia, la dose iniziale raccomandata, indipendentemente dai parametri ematologici, è di 75 mg/m2 per via sottocutanea o endovenosa per 7 giorni al giorno. Dovrebbe essere somministrata una premedicazione per prevenire nausea e vomito.
I successivi cicli di terapia vanno eseguiti ogni 4 settimane. La dose può essere aumentata a 100 mg/m2 in assenza di effetto terapeutico dopo i primi 2 cicli di terapia e in assenza di tossicità (eccetto nausea e vomito). Si consigliano 4-6 cicli di terapia. Se il farmaco risulta totalmente o parzialmente efficace si possono eseguire ulteriori cicli di terapia. Il trattamento può essere continuato finché si osserva una risposta terapeutica.
Prima di cicli ripetuti di terapia, è necessario monitorare la conta assoluta dei neutrofili (ANC) e delle piastrine, nonché le reazioni tossiche (soprattutto a livello renale) e la dose del farmaco deve essere aggiustata di conseguenza.
Se si verifica una diminuzione inspiegabile dei livelli di bicarbonato di sodio<20 мэкв/л при проведении очередного цикла терапии дозу azacitidina dovrebbe essere ridotto del 50%.
In caso di aumento inspiegabile dell’azoto ureico nel sangue (BUN) o della creatinina sierica durante il successivo ciclo di terapia, la dose azacitidina dovrebbero essere ridotti del 50% e mantenuti a questo livello fino a quando questi indicatori non saranno ripristinati ai valori originali (prima del trattamento) o normali.
Dopo il fallimento del trattamento con azacitidina, non ha senso utilizzare la decitabina, poiché quest'ultima non influenza in modo significativo né la durata della remissione né la sopravvivenza globale.
Chemioterapia. Nel trattamento delle MDS “ad alto rischio” vengono utilizzati: chemioterapia secondo il regime “citosar a basso dosaggio” e terapia secondo programmi di leucemia acuta (nell'ambito di studi clinici). Nei pazienti anziani o con gravi patologie concomitanti è indicato l'uso della chemioterapia secondo il regime delle “piccole dosi di Cytazar”.
Basse dosi di Ara-C (livello di evidenza B).
La citarabina viene prescritta 10 mg/m2 (non più di 20 mg) 2 volte al giorno per via sottocutanea per 14-28 giorni. La remissione completa si ottiene nel 15% dei pazienti, parziale nel 20%. Nel 10% si verifica la morte per complicanze infettive dell'agranulocitosi mielotossica.
Terapia secondo programmi per la leucemia acuta.
Secondo NCCN (2.2015)
Non è raccomandato fornire terapia intensiva a pazienti con MDS, incl. rischio elevato al di fuori degli studi clinici. La terapia citostatica secondo il programma per la leucemia acuta viene effettuata con la progressione della malattia, in alcuni casi con RAEB con midollo osseo normo o ipercellulare.
La chemioterapia può essere utilizzata come preparazione al BMT allogenico. (livello di provaC)
.
Terapia di accompagnamento
Principi di adeguata terapia di accompagnamento per le MDS
| Terapia di accompagnamento | Principi di base |
|
Trasfusione |
La trasformazione dei globuli rossi nell'anemia sintomatica avviene principalmente mediante trasfusioni leucofiltrate o piastriniche durante gli episodi emorragici. Le piastrine non devono essere utilizzate di routine nei pazienti con trombocitopenia e senza sanguinamento. L'irradiazione dei componenti del sangue è obbligatoria, da donatori imparentati o HLA-compatibili e nei pazienti pianificati per il trapianto di midollo osseo. |
| Anemia | Si consiglia di determinare il livello iniziale di eritropoietina. La risposta nei pazienti è del 20-30%, entro 1-2 mesi. |
|
Neutropenia/ infezioni |
>50% dei pazienti. Il 30-35% dei pazienti ha neutrofili<1,0x10^9/л или 1.5x109/л, но только у 10% имеются клинически значимые инфекции. Не нужно назначать антибактериальную профилактику всем больным. КСФ увеличивает количество нейтрофилов у 75% больных, но не влияет на общую выживаемость. |
| Trombocitopenia | 25-50% dei pazienti. Gli agenti trombopoietici non influenzano significativamente la necessità di emocomponenti (TPO, MGDF, IL-11) |
Eriropoietine.
Eritropoietina (EPO)- il principale regolatore dell'eritropoiesi - stimola la formazione di globuli rossi dalle cellule progenitrici tardive e aumenta la resa dei reticolociti dal midollo osseo.
La terapia con EPO nel gruppo generale di pazienti con MDS è efficace solo nel 15-30% dei casi e aumenta al 70% quando prescritta per le seguenti indicazioni: emoglobina<100 г/л, эндогенный ЭПО <200 ЕД/мл, низкая или промежуточная I группа риска по IPSS. (Livello di evidenza A)
Terapia iniziale(fase di correzione). Quando somministrata per via endovenosa, la dose iniziale è di 40 UI/kg di peso corporeo 3 volte a settimana (la dose più alta non deve superare 720 UI/kg di peso corporeo a settimana).
Terapia di mantenimento. Per mantenere l'ematocrito al 30-35%, la dose deve essere inizialmente ridotta alla metà della dose dell'iniezione precedente. Successivamente, la dose di mantenimento viene scelta individualmente, ad intervalli di una o due settimane.
Al fine di ridurre il numero di iniezioni e migliorare la qualità della vita, è possibile utilizzare EPO a rilascio prolungato 300 mcg per via sottocutanea una volta ogni 3 settimane.
Trattamento della trombocitopenia (livello di evidenza)B).
I farmaci stimolanti la trombopoiesi vengono utilizzati:
Romiplostim AMG 531 (pepticorpo TPO-R);
· Eltrombopag (agonista TPO-R-GSK);
Lonifarnib, Tipifarnib.
Romiplostim- promuove la differenziazione dei megacariociti e delle piastrine allo stesso modo della trombopoietina endogena. È un analogo della trombopoietina, che porta ad un aumento della produzione di piastrine. Somministrato per via sottocutanea. La dose iniziale è di 1 mcg/kg di peso corporeo reale, settimanalmente, fino a quando la conta piastrinica si stabilizza a 50 x 10 9 / L o più per almeno 4 settimane senza aggiustamento della dose.
Terapia chelante (livello di evidenza B).
Trattamento standard per l'avvelenamento da metalli pesanti. Il metodo di chelazione è stato sviluppato negli anni '50. per il trattamento dell'avvelenamento da metalli pesanti: piombo, ferro, mercurio e rame. Il metodo viene utilizzato per trattare pazienti affetti da MDS con sovraccarico di ferro. L'indicazione standard per iniziare la terapia chelante è un aumento dei livelli di ferritina superiore a 1000 mcg/L.
Deferoxamina (Livello di evidenza B). La dose media giornaliera è di 20-40 mg/kg. La dose iniziale media è di 500 mg. La terapia è considerata efficace se le concentrazioni di ferritina sierica sono vicine ai valori normali (meno di 300 μg/L).
Deferasirox. La dose iniziale raccomandata è di 20 mg, assunta per via orale una volta al giorno.
Supporto trasfusionale.
Le indicazioni per la terapia trasfusionale sono determinate principalmente dalle manifestazioni cliniche individualmente per ciascun paziente, tenendo conto dell'età, delle malattie concomitanti, della tollerabilità della chemioterapia e dello sviluppo di complicanze nelle fasi precedenti del trattamento.
Gli indicatori di laboratorio per determinare le indicazioni hanno un valore ausiliario, principalmente per valutare la necessità di trasfusioni profilattiche di concentrato piastrinico.
Le indicazioni per le trasfusioni dipendono anche dal tempo trascorso dopo un ciclo di chemioterapia: viene presa in considerazione la prevista diminuzione degli indicatori nei prossimi giorni.
Massa/sospensione dei globuli rossi (livello di evidenza)D) :
· Non è necessario aumentare i livelli di emoglobina finché le normali riserve e i meccanismi di compensazione sono sufficienti a soddisfare il fabbisogno di ossigeno dei tessuti;
· Esiste una sola indicazione per la trasfusione di mezzi contenenti globuli rossi per l'anemia cronica: anemia sintomatica (manifestata da tachicardia, mancanza di respiro, angina pectoris, sincope, depressione denovo o sopraslivellamento del tratto ST);
· Un livello di emoglobina inferiore a 30 g/l è un'indicazione assoluta alla trasfusione di globuli rossi;
· In assenza di malattie scompensate del sistema cardiovascolare e dei polmoni, i livelli di emoglobina possono essere indicazioni per la trasfusione profilattica di globuli rossi nell'anemia cronica:
Le trasfusioni di globuli rossi a pazienti con MDS dovrebbero essere effettuate con il monitoraggio dinamico del metabolismo del ferro. Il sovraccarico di ferro conseguente a trasfusioni multiple di sangue costituisce un'indicazione alla terapia chelante
Concentrato piastrinico per MDS (livello di evidenza)D). :
· Se il livello delle piastrine scende al di sotto di 30 x 10 9 / l, viene eseguita una trasfusione di piastrine in aferesi per mantenere il loro livello non inferiore a 30-50 x 10 9 / l, soprattutto nei primi 10 giorni del ciclo.
· Se esiste un rischio elevato di complicanze emorragiche (età superiore a 60 anni, iperleucocitosi (più di 10x10 9 /l), aumento del livello di creatinina superiore a 140 µmol/l), è necessario mantenere un livello piastrinico superiore a 50x10 9 /l.
Plasma fresco congelato (livello di evidenza)D) :
· Le trasfusioni di FFP vengono eseguite in pazienti con sanguinamento o prima di interventi invasivi;
· I pazienti con un INR pari a ³2,0 (per interventi neurochirurgici ³1,5) sono considerati candidati alla trasfusione di FFP quando si pianificano procedure invasive.
Terapie farmacologiche erogate in regime ambulatoriale:
− elenco dei medicinali essenziali con indicazione della forma di rilascio (aventi una probabilità di utilizzo del 100%):
Farmaci antineoplastici e immunosoppressori:
- ciclosporina*, 100 mg, 50 mg, capsule;
- romiplostim 250 mg, 500 mg polvere per soluzione per somministrazione sottocutanea.
− elenco dei medicinali aggiuntivi indicante la forma di rilascio (probabilità di utilizzo inferiore al 100%):
· deferoxamina*, 500 mg;
· filgrastim, soluzione iniettabile 0,3 mg/ml, 1 ml;
Agenti antibatterici:
Azitromicina, compressa/capsula, 500 mg;
· amoxicillina/acido clavulanico, compressa rivestita con film, 1000 mg;
· moxifloxacina, compressa, 400 mg;
Ofloxacina, compressa, 400 mg;
· compressa di ciprofloxacina, 500 mg;
· metronidazolo, compressa, 250 mg;
Levofloxacina, compressa, 500 mg.
· anidulafungina, polvere liofilizzata per soluzione iniettabile, 100 mg/flacone;
voriconazolo, compressa, 50 mg;
· clotrimazolo, soluzione per uso esterno 1% 15ml;
Fluconazolo, capsula/compressa 150 mg.
· aciclovir, compressa, 400 mg, crema per uso esterno, 5% - 5,0;
· valganciclovir, compressa, 450 mg;
Famciclovir compresse da 500 mg.
· sulfametossazolo/trimetoprim, compressa da 480 mg.
Soluzioni utilizzate per correggere i disturbi dell'equilibrio idrico, elettrolitico e acido-base:
· destrosio, soluzione per infusione al 5% 250ml;
· cloruro di sodio, soluzione per infusione 0,9% 500 ml.
· eparina, soluzione iniettabile 5000 UI/ml - 5 ml; (per il lavaggio del catetere), gel in tubo 100000IU 50g
· rivaroxaban, compressa.
acido tranexamico, capsula/compressa 250 mg;
· enoxaparina, soluzione iniettabile in siringhe da 4000 anti-Xa UI/0,4 ml, 8000 anti-Xa UI/0,8 ml;
· ambroxolo, soluzione per somministrazione orale e inalazione, 15 mg/2 ml, 100 ml;
· atenololo, compressa 25 mg;
· drotaverina, compressa 40 mg;
Lisinopril, compressa da 5 mg;
· omeprazolo, capsula 20 mg;
Prednisolone, compressa, 5 mg;
· smectite diottaedrica, polvere per preparazione della sospensione per somministrazione orale 3,0 g;
· torasemide, compressa 10 mg;
· fentanil, sistema terapeutico transdermico 75 mcg/h; (per il trattamento del dolore cronico nei pazienti oncologici)
Trattamento farmacologico fornito a livello ospedaliero:
elenco dei medicinali essenziali con indicazione della forma di rilascio (aventi una probabilità di utilizzo del 100%):
- decitabina 50 mg, liofilizzato per la preparazione della soluzione per infusione;
- citarabina, fl. 100 mg, concentrato per soluzione;
- azacitidina*100 mg, flacone, concentrato per soluzione;
- ciclosporina*, capsula 50 mg, capsula 100 mg;
- romiplostim 250 mg, 500 mg, polvere per soluzione per somministrazione sottocutanea;
Medicinali che indeboliscono l’effetto tossico dei farmaci antitumorali:
· deferasirox*, tab. 250 mg;
- deferoxamina*, 500 mg;
· filgrastim, soluzione iniettabile 0,3 mg/ml, 1 ml;
· ondansetron, soluzione iniettabile 8 mg/4 ml.
Agenti antibatterici:
· azitromicina, compressa/capsula, 500 mg, polvere liofilizzata per la preparazione di soluzione per infusione endovenosa, 500 mg;
· amikacina, polvere per iniezione, 500 mg/2 ml o polvere per soluzione iniettabile, 0,5 g;
· amoxicillina/acido clavulanico, compressa rivestita con film, 1000 mg, polvere per soluzione per somministrazione endovenosa e intramuscolare 1000 mg+500 mg;
· vancomicina, polvere/liofilizzato per soluzione per infusione 1000 mg;
· gentamicina, soluzione iniettabile 80 mg/2 ml 2 ml;
· imipinem, cilastatina polvere per soluzione per infusione, 500 mg/500 mg;
Levofloxacina, compressa, 500 mg, soluzione per infusione 500 mg/100 ml;
linezolid, soluzione per infusione 2 mg/ml;
· meropenem, liofilizzato/polvere per soluzione iniettabile 1,0 g;
· moxifloxacina, compressa 400 mg, soluzione per infusione 400 mg/250 ml;
· ofloxacina, compressa 400 mg, soluzione per infusione 200 mg/100 ml;
· piperacillina, tazobactam polvere per soluzione iniettabile 4,5 g;
tigeciclina*, polvere liofilizzata per soluzione iniettabile 50 mg/flacone;
Ticarcillina/acido clavulanico, polvere liofilizzata per la preparazione della soluzione per infusione 3000 mg/200 mg;
cefepime, polvere per soluzione iniettabile 500 mg, 1000 mg;
· cefoperazone, sulbactam polvere per soluzione iniettabile 2 g;
· ciprofloxacina, compressa 500 mg, soluzione per infusione 200 mg/100 ml - 100 ml;
· eritromicina, compressa 250 mg;
· ertapenem liofilizzato, per la preparazione di una soluzione per iniezioni endovenose e intramuscolari 1 g;
Farmaci antifungini:
· amfotericina B*, polvere liofilizzata per soluzione iniettabile, 50 mg/flacone;
· anidulofungina, polvere liofilizzata per soluzione iniettabile, 100 mg/flacone;
voriconazolo, compressa, 50 mg, polvere per soluzione per infusione 200 mg/flacone;
· itraconazolo soluzione orale 10 mg/ml 150,0;
· caspofungin, liofilizzato per la preparazione della soluzione per infusione 50 mg;
· clotrimazolo, crema per uso esterno 1% 30g, 15ml;
· colistimetato di sodio*, liofilizzato per la preparazione della soluzione per infusione, 1 milione di unità/flacone;
· micafungin, polvere liofilizzata per la preparazione di soluzione iniettabile 50 mg, 100 mg;
· fluconazolo, capsula/compressa 150 mg, soluzione per infusione 200 mg/100 ml - 100 ml.
Farmaci antivirali:
· aciclovir, compressa, 400 mg, crema per uso esterno, 5% - 5,0, polvere per soluzione per infusione, 250 mg;
· valaciclovir, compressa, 500 mg;
· valganciclovir, compressa, 450 mg;
· ganciclovir*, liofilizzato per soluzione per infusione 500 mg;
Famciclovir, compresse, 500 mg n. 14.
Medicinali usati per la pneumocistosi:
· sulfametossazolo/trimetoprim, compressa 480 mg, concentrato per soluzione per infusione (80 mg+16 mg)/ml, 5 ml.
Ulteriori farmaci immunosoppressori:
· desametasone, soluzione iniettabile 4 mg/ml 1 ml;
· metilprednisolone, compressa, 16 mg;
· metilprednisolone, soluzione iniettabile, 250 mg;
· prednisolone, soluzione iniettabile 30 mg/ml 1 ml, compressa, 5 mg.
Soluzioni utilizzate per correggere i disturbi dell'equilibrio idrico, elettrolitico e acido-base, nutrizione parenterale:
· albumina, soluzione per infusione 10% - 100 ml, 20% - 100 ml;
· acqua per preparazioni iniettabili, soluzione iniettabile 5 ml;
· destrosio, soluzione per infusione al 5% - 250ml, 500ml;
· destrosio, soluzione iniettabile al 40% - 10 ml, 20 ml;
· cloruro di potassio, soluzione per somministrazione endovenosa 40 mg/ml, 10 ml;
· gluconato di calcio, soluzione iniettabile al 10%, 5 ml;
· cloruro di calcio, soluzione iniettabile al 10% 5 ml;
· solfato di magnesio, soluzione iniettabile al 25% 5 ml;
· mannitolo, soluzione iniettabile 15% -200,0;
· cloruro di sodio, soluzione per infusione 0,9% - 250 ml, 500 ml;
· soluzione per infusione di cloruro di sodio, cloruro di potassio, acetato di sodio in flaconi da 200 ml, 400 ml;
· soluzione per infusione di cloruro di sodio, cloruro di potassio, acetato di sodio 200 ml, 400 ml;
· soluzione per infusione di cloruro di sodio, cloruro di potassio, bicarbonato di sodio 400ml;
L-alanina, L-arginina, glicina, L-istidina, L-isoleucina, L-leucina, L-lisina cloridrato, L-metionina, L-fenilalanina, L-prolina, L-serina, L-treonina, L-triptofano , L-tirosina, L-valina, sodio acetato triidrato, sodio glicerofosfato pentiidrato, potassio cloruro, magnesio cloruro esaidrato, glucosio, calcio cloruro diidrato, emulsione di oli di oliva e di soia miscela per inf.: contenitori a tre camere 2 l
· amido idrossietilico (pentastarco), soluzione per infusione al 6% 500 ml;
· complesso di aminoacidi, emulsione per infusione contenente una miscela di oli di oliva e di soia in rapporto 80:20, una soluzione di aminoacidi con elettroliti, una soluzione di destrosio, con un contenuto calorico totale di 1800 kcal Contenitore a tre sezioni da 1.500 ml .
Medicinali utilizzati per la terapia intensiva (farmaci cardiotonici per il trattamento dello shock settico, miorilassanti, vasopressori e anestetici):
· aminofillina, soluzione iniettabile 2,4%, 5 ml;
· amiodarone, soluzione iniettabile, 150 mg/3 ml;
· atracurio besilato, soluzione iniettabile, 25 mg/2,5 ml;
· atropina, soluzione iniettabile, 1 mg/ml;
· diazepam, soluzione per uso intramuscolare ed endovenoso 5 mg/ml 2 ml;
· dobutamina*, soluzione iniettabile 250 mg/50,0 ml;
· dopamina, soluzione/concentrato per la preparazione della soluzione iniettabile 4%, 5 ml;
· insulina semplice;
· ketamina, soluzione iniettabile 500 mg/10 ml;
· morfina, soluzione iniettabile 1% 1 ml;
· norepinefrina*, soluzione iniettabile 20 mg/ml 4,0;
· pipecuronio bromuro, polvere liofilizzata per preparazioni iniettabili 4 mg;
· propofol, emulsione per somministrazione endovenosa 10 mg/ml - 20 ml, 50 ml;
· rocuronio bromuro, soluzione per somministrazione endovenosa 10 mg/ml, 5 ml;
· tiopentale sodico, polvere per la preparazione della soluzione per somministrazione endovenosa 500 mg;
· fenilefrina, soluzione iniettabile 1% 1ml;
immunoglobulina umana normale, soluzione per infusione;
· epinefrina, soluzione iniettabile 0,18% 1 ml.
Medicinali che influenzano il sistema di coagulazione del sangue:
· acido aminocaproico, soluzione al 5% -100 ml;
· complesso coagulante anti-inibitorio, polvere liofilizzata per la preparazione della soluzione iniettabile, 500 UI;
· eparina, soluzione iniettabile 5000 UI/ml - 5 ml, gel in tubo 100000 UI 50g;
· spugna emostatica, misura 7*5*1, 8*3;
· nadroparina, soluzione iniettabile in siringhe preriempite, 2850 UI anti-Xa/0,3 ml, 5700 UI anti-Xa/0,6 ml;
· enoxaparina, soluzione iniettabile in siringhe da 4000 anti-Xa UI/0,4 ml, 8000 anti-Xa UI/0,8 ml.
Altri medicinali:
· ambroxolo, soluzione iniettabile, 15 mg/2 ml, soluzione per somministrazione orale e inalazione, 15 mg/2 ml - 100 ml, soluzione per somministrazione orale e inalazione, 15 mg/2 ml, 100 ml;
· amlodipina, compressa/capsula 5 mg;
· atenololo, compressa 25 mg;
· acetilcisteina, polvere per soluzione per somministrazione orale, 3 g;
· bupivacaina, soluzione iniettabile 5 mg/ml, 4 ml;
· eparina gel in tubo 100.000 unità 50 g;
· desametasone, collirio 0,1% 8 ml;
Difenidramina, soluzione iniettabile 1% 1 ml;
· drotaverina, soluzione iniettabile al 2%, 2 ml;
· soluzione normale di immunoglobulina umana per somministrazione endovenosa 50 mg/ml - 50 ml;
· captopril, compressa 50 mg;
· ketoprofene, soluzione iniettabile 100 mg/2ml;
lattulosio, sciroppo 667 g/l, 500 ml;
· cloramfenicolo, sulfadimetossina, metiluracile, trimecaina unguento per uso esterno 40g;
· lidocaina, soluzione iniettabile, 2%, 2 ml;
Lisinopril, compressa da 5 mg;
· metiluracile, pomata ad uso topico in tubo 10% 25g;
· metronidazolo, gel dentale 20g;
· nafazolina gocce nasali 0,1% 10 ml;
· nicergolina, liofilizzato per la preparazione della soluzione iniettabile 4 mg;
· omeprazolo, capsula 20 mg, polvere liofilizzata per la preparazione della soluzione iniettabile 40 mg;
· omeprazolo, polvere liofilizzata per la preparazione della soluzione iniettabile da 40 mg;
· iodio-povidone, soluzione per uso esterno 1 l;
· procaina, soluzione iniettabile 0,5%, 10 ml;
· salbutamolo, soluzione per nebulizzatore 5 mg/ml-20 ml;
· smectitediottaedrico, polvere per la preparazione della sospensione per somministrazione orale 3,0 g;
· spironolattone, capsula 100 mg;
· tobramicina, collirio 0,3% 5ml
· torasemide, compressa 10 mg;
tramadolo, capsule 50, 100 mg;
tramadolo, compresse 50 mg
· famotidina, polvere liofilizzata per la preparazione di soluzione iniettabile 20 mg;
· fenobarbital, compressa 100 mg;
· fentanil, sistema terapeutico transdermico 75 mcg/h (per il trattamento del dolore cronico nei pazienti oncologici);
· acido folico, compressa, 5 mg;
· furosemide, soluzione iniettabile 1% 2 ml;
· cloramfenicolo, sulfadimetossina, metiluracile, trimecaina unguento per uso esterno 40g;
· clorexidina, soluzione 0,05% 100ml;
· cloropiramina, soluzione iniettabile 20 mg/ml 1 ml.
Terapie farmacologiche erogate in fase di emergenza: non viene effettuato.
Altri tipi di trattamento:
Altri tipi di cure erogate in regime ambulatoriale: non applicare.
Altri tipi di trattamento forniti a livello ospedaliero:
Trapianto di midollo osseo allogenico per MDS (livello di evidenza B).
Il trapianto allogenico di midollo osseo è l’unico trattamento radicale per i pazienti con sindrome mielodisplastica. È stato notato un aumento della sopravvivenza globale nel gruppo di pazienti con un regime di condizionamento standard.La sopravvivenza globale e quella libera da eventi non dipendono dall'età, dal tipo di donatore, dalla compatibilità HLA, dalla fonte delle cellule staminali, dal numero di blasti prima del trapianto. dopo che l'alloBMT viene raggiunto dal 60-70% dei pazienti con MDS, tuttavia, di norma, le remissioni sono di breve durata.
La fonte delle HSC per alloBMT è un donatore correlato HLA identico, ma i risultati del trapianto da un donatore non correlato HLA identico sono paragonabili ai risultati del BMT da un donatore correlato.
In assenza di donatori HLA compatibili, un donatore correlato aploidentico può essere considerato una fonte alternativa di CSE (livello di provaD).
Per i pazienti del gruppo “a basso rischio”. Il BMT è indicato in caso di mancata risposta alla terapia e persistenza della dipendenza trasfusionale (trasfusione di almeno 1 dose di globuli rossi più di ogni 8 settimane per 4 mesi).
Per i pazienti del gruppo “ad alto rischio” alloBMT Si consiglia di effettuarlo immediatamente dopo aver ottenuto una risposta completa o parziale alla terapia.
Altri tipi di trattamento forniti durante le cure mediche di emergenza: non applicare.
Intervento chirurgico:
Intervento chirurgico erogato in regime ambulatoriale: non viene effettuato.
Intervento chirurgico erogato in regime di ricovero: Se si sviluppano complicazioni infettive e sanguinamenti potenzialmente letali, i pazienti possono essere sottoposti a interventi chirurgici per indicazioni di emergenza.
Ulteriore gestione:
1) D-registrazione con un ematologo, controllo emocromo + piastrine una volta ogni 14 giorni;
2) Trasfusione di emocomponenti secondo rigorose indicazioni cliniche, quanto più limitate possibile;
3) Controllare la ferritina (dopo ogni 10 trasfusioni), fare un esame del sangue (ferritina).
Indicatori di efficacia del trattamento:
| Categoria | Criteri di risposta |
| Risposta completa | Nel midollo osseo ≤5% dei mieloblasti con |
| Nessuna displasia. | |
|
Parametri del sangue periferico: L'emoglobina è superiore a 11 g/dl Piastrine > = 100 x 10 9/L Neutrofili > = 1x 10 9/L Celle esplosive = 0% x 10 9/L |
|
| Risposta parziale | Nel midollo osseo il numero dei blasti è diminuito del 50% rispetto al livello iniziale, ma rimane superiore al 5% |
| Remissione completa del midollo osseo |
Il midollo osseo contiene ≤ 5% di mieloblasti e diminuisce di oltre il 50% dopo il trattamento. Nel sangue periferico: si è avuta la stessa risposta. |
| Remissione incompleta | Mancato raggiungimento di una risposta incompleta ma nessuna evidenza di progressione dopo 8 settimane. |
| Fallimento del trattamento | Morte durante il trattamento dovuta a progressione della malattia, aumento della citopenia, aumento del numero di blasti nel midollo osseo, progressione della malattia. |
| Recidiva dopo risposta completa e parziale |
almeno 1 dei seguenti: Rilevazione di blasti nel midollo osseo>50% dopo la remissione diminuzione dell'emoglobina >150 g/l e aumento della dipendenza dalle trasfusioni. |
| Risposta citogenetica |
Scomparsa delle anomalie cromosomiche, senza la comparsa di nuove. Risposta incompleta: Riduzione delle anomalie cromosomiche di almeno il 50%. |
Farmaci (principi attivi) utilizzati nel trattamento
| Spugna emostatica |
| Azacitidina |
| Azitromicina |
| Albumina umana |
| Ambroxolo |
| Amikacina |
| Acido aminocaproico |
| Aminoacidi per nutrizione parenterale+Altri medicinali (Emulsioni di grassi + Destrosio + Multiminerale) |
| Aminofillina |
| Amiodarone |
| Amlodipina |
| Amoxicillina |
| Amfotericina B |
| Anidulafungina |
| Complesso coagulante antiinibitorio |
| Atenololo |
| Atracurium besilato |
| Atropina |
| Acetilcisteina |
| Aciclovir |
| Bupivacaina |
| Valaciclovir |
| Valganciclovir |
| Vancomicina |
| Acqua per preparazioni iniettabili |
| Voriconazolo |
| Ganciclovir |
| Gentamicina |
| Eparina sodica |
| Amido idrossietilico |
| Desametasone |
| Destrosio |
| Deferasirox |
| Deferoxamina |
| Decitabina |
| Diazepam |
| Difenidramina |
| Dobutamina |
| Dopamina |
| Drotaverina (Drotaverina) |
| Imipenem |
| Antitimociti immunoglobulinici |
| Immunoglobulina umana normale (IgG+IgA+IgM) (Immunoglobulina umana normale (IgG+IgA+IgM)) |
| Immunoglobulina umana normale |
| Itraconazolo |
| Cloruro di potassio (Cloruro di potassio) |
| Gluconato di calcio |
| Captopril |
| Caspofungina |
| Ketamina |
| Ketoprofene |
| Acido clavulanico |
| Clotrimazolo |
| Colistimetato di sodio |
| Complesso di aminoacidi per nutrizione parenterale |
| Concentrato piastrinico (CT) |
| Lattulosio |
| Levofloxacina |
| Lenalidomide |
| Lidocaina |
| Lisinopril |
| Linezolid |
| Solfato di magnesio |
| Mannitolo |
| Meropenem |
| Metilprednisolone |
| Metiluracile (diossometiltetraidropirimidina) |
| Metronidazolo |
| Micafungina |
| Moxifloxacina |
| Morfina |
| Nadroparina calcio |
| Acetato di sodio |
| Idrocarbonato di sodio |
| Cloruro di sodio |
| Nafazolina |
| Nicergolina |
| Noradrenalina |
| Omeprazolo |
| Ondansetrone |
| Ofloxacina |
| Bromuro di pipecuronio |
| Piperacillina |
| Plasma fresco congelato |
| Iodio povidone |
| Prednisolone |
| Procaina |
| Propofol |
| Rivaroxaban |
| Rocuronio bromuro |
| Romiplostim |
| Salbutamolo |
| Smectite diottaedrica |
| Miscele per nutrizione enterale |
| Spironolattone |
| Sulfametossazolo |
| Tazobactam |
| Tigeciclina |
| Ticarcillina |
| Tiopentale sodico |
| Tobramicina |
| Torasemide |
| Tramadolo |
| Acido tranexamico |
| Trimecaina |
| Trimetoprim |
| Famotidina |
| Famciclovir |
| Fenilefrina |
| Fenobarbital |
| Fentanil |
| Fentanil |
| Filgrastim |
| Fluconazolo |
| Acido folico |
| Furosemide |
| Cloramfenicolo |
| Clorexidina |
| Clorexidina |
| Cloropiramina |
| Cefepime |
| Cefoperazone |
| Ciclosporina |
| Ciprofloxacina |
| Citarabina |
| Eltrombopag |
| Enoxaparina sodica |
| Epinefrina |
| Eritromicina |
| Massa dei globuli rossi |
| Sospensione eritrocitaria |
| Ertapenem |
Gruppi di farmaci secondo ATC utilizzati nel trattamento
Ricovero ospedaliero
Indicazioni per il ricovero:
Indicazioni per il ricovero d'urgenza:
· sindrome mielodisplastica di nuova diagnosi;
· sindromi emorragiche e anemiche con manifestazioni cliniche pronunciate;
neutropenia febbrile.
Indicazioni per il ricovero programmato:
· Effettuazione di terapia immunosoppressiva con ATG;
· Effettuazione della chemioterapia;
· Trapianto allogenico di cellule staminali emopoietiche.
Prevenzione
Azioni preventive: NO.
Informazione
Fonti e letteratura
- Verbali delle riunioni del Consiglio di esperti dell'RCHR del Ministero della Salute della Repubblica del Kazakistan, 2015
- Riferimenti: 1. Rete scozzese di linee guida intercollegiate (SIGN). SIGN 50: un manuale per gli sviluppatori di linee guida. Edimburgo: segno; 2014. (Pubblicazione SIGN n. 50). . Disponibile dall'URL: http://www.sign.ac.uk. 2. Volkova M.A. Oncoematologia clinica. – M.: Medicina, 2007. – 1120 p. 3. Linee guida per la pratica clinica del NCCN in oncologia (linee guida del NCCN) Sindromi mielodisplastiche. www.nccn.org 4. Greenberg P, Cox C, LeBeau MM et al. Sistema di punteggio internazionale per la valutazione della prognosi nelle sindromi mieloblastiche // Sangue. – 1997. – Vol. 89. – P. 2088–2089. 5. Ematologia; L'ultimo libro di consultazione. Sotto la direzione generale del Dottore in Scienze Mediche. Il professor K.M. Abdulkadyrova. Mosca: Casa editrice Eksmo; San Pietroburgo: Casa editrice Sova, 2004; 601-683 6. Panoramica sulla sindrome mielodisplastica di Kurzrock R. // SeminHematol. – 2002. – Vol. 39. – P. 18–25. 7. Carr SE, Halliday V. Indagare sull'uso della dieta neutropenica: un'indagine sui dietisti del Regno Unito. Dieta J Hum Nutr. 28 agosto 2014. 8. Boeckh M. Dieta neutropenica: buona pratica o mito? Trapianto di midollo sanguigno Biol. settembre 2012; 18(9):1318-9. 9. Trifilio, S., Helenowski, I., Giel, M. et al. Mettere in discussione il ruolo di una dieta neutropenica dopo il trapianto di cellule staminali emopoietiche. Trapianto di midollo sanguigno Biol. 2012; 18: 1387–1392 10. Greenberg PL. Sindromi mielodisplastiche: conseguenze del sovraccarico di ferro e attuali terapie chelanti. J Natl Compr CancNetw 2006;4:91-96 11. Jensen PD, Jensen FT, Christensen T, et al. Relazione tra danno epatocellulare e sovraccarico di ferro trasfusionale prima e durante la chelazione del ferro con desferrioxamina: uno studio in pazienti adulti con anemie acquisite. Sangue 2003;101:91-96. 12. Jensen PD, Jensen FT, Christensen T et al. Valutazione del ferro miocardico mediante risonanza magnetica durante la terapia ferrochelante con deferrioxamina: indicazione di una stretta relazione tra contenuto di ferro miocardico e pool di ferro chelabile. Blood 2003;101:4632-4639 13. Passweg JR, Giagounidis AA, Simcock M, et al. Terapia immunosoppressiva per pazienti con sindrome mielodisplastica: uno studio prospettico randomizzato multicentrico di fase III che confronta la globulina antitimocitaria più ciclosporina con la migliore terapia di supporto - SAKK 33/99. J Clin Oncol 2011; 29:303-309. 14. Stadler M, Germing U, Kliche KO, et al. Uno studio prospettico, randomizzato, di fase II sulla globulina antitimocitaria di cavallo rispetto alla globulina antitimocitaria di coniglio come terapia immunomodulante in pazienti con sindromi mielodisplastiche a basso rischio. Leucemia 2004; 18:460-465. 15. Saunthararajah Y, Nakamura R, Nam JM, et al. HLA-DR15 (DR2) è sovrarappresentato nella sindrome mielodisplastica e nell'anemia aplastica e predice una risposta all'immunosoppressione nella sindrome mielodisplastica. Blood 2002;100:1570-1574 16. Sloand EM, Wu CO, Greenberg P, et al. Fattori che influenzano la risposta e la sopravvivenza nei pazienti con mielodisplasia trattati con terapia immunosoppressiva. J Clin Oncol 2008;26:2505-2511 17. Jadersten M, Saft L, Pellagatti A, et al. Eterogeneità clonale nella sindrome 5q-: i progenitori che esprimono p53 prevalgono durante il trattamento con lenalidomide e si espandono con la progressione della malattia. Haematologica 2009;94:1762-1766 18. Mallo M, Del Rey M, Ibanez M, et al. Risposta alla lenalidomide nelle sindromi mielodisplastiche con del(5q): influenza della citogenetica e delle mutazioni. Br J Haematol 2013;162:74-86 19. Greenberg PL, Lee SJ, Advani R, et al. Mitoxantrone, etoposide e citarabina con o senza valspodar in pazienti con leucemia mieloide acuta recidivante o refrattaria e sindrome mielodisplastica ad alto rischio: uno studio di fase III (E2995). J Clin Oncol 2004;22:1078-1086 20. McClune B, Weisdorf D, DiPersio J, et al. Trapianto di cellule staminali ematopoietiche non mieloablative in pazienti anziani con leucemia mieloide acuta e sindromi mielodisplastiche: risultati del Centro internazionale per la ricerca sui trapianti di sangue e midollo (CIBMTR). Blood 2008;112: 21. Lubbert M, Bertz H, Ruter B, et al. Trattamento non intensivo con 5-aza-2"-deossicitidina (DAC) a basso dosaggio prima di SCT ematico allogenico di pazienti affetti da MDS/AML più anziani. Bone Marrow Transplant 2009;44:585-588 22. McClune BL, Weisdorf DJ, Pedersen TL, et al. Effetto dell'età sull'esito del trapianto di cellule ematopoietiche a intensità ridotta per pazienti anziani con leucemia mieloide acuta in prima remissione completa o con sindrome mielodisplastica. J Clin Oncol 2010;28:1878-1887. 23. Kantarjian H, Oki Y, Garcia-Manero G, et al. Risultati di uno studio randomizzato su 3 programmi di decitabina a basso dosaggio nella sindrome mielodisplastica ad alto rischio e nella leucemia mielomonocitica cronica. Blood 2007;109:52-57. 24. Saba H, Lubbert M , Wijermans PW. Tassi di risposta degli studi di fase 2 e fase 3 sulla decitabina (DAC) in pazienti con sindromi mielodisplastiche (MDS). Blood 2005;106: 25. Saba H, Rosenfeld C, Issa J, et al. Primo rapporto del Studio nordamericano di fase III sulla decitabina nella sindrome mielodisplastica avanzata (MDS) Blood 2004;104:Abstract 67. 26. Fili C, Malagola M, Follo MY, et al. Studio prospettico di fase II su azacitidina per 5 giorni per il trattamento di pazienti sintomatici e/o non responsivi all'eritropoietina affetti da sindromi mielodisplastiche a basso/rischio T-1 IN. Clin Cancer Res 2013; 19:3297–3308. 27. Gore SD, Fenaux P, Santini V, et al. Un'analisi multivariata riguarda la relazione tra risposta e sopravvivenza tra i pazienti con sindromi mielodisplastiche ad alto rischio trattati con azacitidina o regimi di cura convenzionali nello studio randomizzato AZA-001. Ematologica 2013; 98:1067–1072. 28. Silverman LR, Demakos EP, Peterson BL, et al. Studio randomizzato e controllato sull'azacitidina in pazienti con sindrome mielodisplastica: uno studio sul gruppo di cancro e leucemia B. J Clin Oncol 2002;20:2429-2440. 29. Della Porta MG, Alessandrino EP, Bacigalupo A, et al. Fattori predittivi per l'esito del trapianto allogenico in pazienti con MDS stratificati secondo l'IPSS-R revisionato. Sangue 2014; 123:2333–2342. 30. Damaj G, Duhamel A, Robin M, et al. Impatto dell'azacitidina prima del trapianto allogenico di cellule staminali per le sindromi mielodisplastiche: uno studio della Société Francaise de Greffe de Moelle et de Therapie-Cellulaire e del Groupe-Francophone des Myelodysplasies. J Clin Oncol 2012;30:4533-4540 31. Scott BL, Sandmaier BM, Storer B, et al. Trapianto allogenico mieloablativo vs non mieloablativo per pazienti con sindrome mielodisplastica o leucemia mieloide acuta con displasia multilineare: un'analisi retrospettiva. Leukemia 006;20:128-135 32. Peter L. Greenberg, Guillermo Garcia-Manero, Michael Moore, Lloyd Damon, Gail Roboz, Kuolung Hu, Allen S. Yang e Janet Franklin Uno studio randomizzato e controllato su romiplostim in pazienti con bassa - o sindrome mielodisplastica a rischio intermedio trattati con decitabina. Linfoma di Leuk. 2013 Feb;54(2):321-8 33. Mixue Xie, Qi Jiang, Yanhui Xie Confronto tra decitabina e azacitidina per il trattamento della sindrome mielodisplastica: una meta-analisi con 1392 partecipantiClin Lymphoma Myeloma Leuk.2015 Jan;15(1 ):22-8 34. Tricot G, Boogaerts MA. Il ruolo della chemioterapia aggressiva nel trattamento delle sindromi mielodisplastiche. Br J Ematolo. 1986 Jul;63(3):477-83 35. Aul C1, Schneider W. Il ruolo della citosina arabinoside a basso dosaggio e della chemioterapia aggressiva nelle sindromi mielodisplastiche avanzate. Cancro. 1 novembre 1989;64(9):1812-8. 36. Hellstrom-Lindberg E. Efficacia dell'eritropoietina nelle sindromi mielodisplastiche: una meta-analisi di 205 pazienti da 17 studi. Br J Haematol 1995;89:67-71 37. Tamura H, Ogata K, Luo S, et al. Livelli plasmatici di trombopoietina (TPO) ed espressione del recettore TPO sulle piastrine in pazienti con sindromi mielodisplastiche. Br J Haematol 1998;103:778-784 38. Harel S, Cherait A, Berthon C, Willekens C, Park S, Rigal M, Brechignac S, Thépot S, Quesnel B, Gardin C, Adès L, Fenaux P, Braun T Esiti di pazienti con sindrome mielodisplastica (MDS) ad alto rischio e leucemia mielomonocitica cronica avanzata (CMML) trattati con decitabina dopo fallimento di azacitidina. Leuk Ris. 2015 maggio;39(5):501-4. doi: 10.1016/j.leukres.2015.02.004. Epub 2015, 16 febbraio.
Informazione
Elenco degli sviluppatori di protocolli con informazioni sulla qualifica:
1) Kemaikin Vadim Matveevich - Candidato di scienze mediche, JSC "Centro scientifico nazionale di oncologia e trapianti", capo del dipartimento di oncoematologia e trapianto di midollo osseo.
2) Anton Anatolyevich Klodzinsky - Candidato di scienze mediche, Centro scientifico nazionale di oncologia e trapianti JSC, ematologo presso il Dipartimento di oncoematologia e trapianto di midollo osseo.
3) Ramazanova Raigul Mukhambetovna - Dottore in scienze mediche, professore alla JSC "Kazakh Medical University of Continuing Education", capo del corso di ematologia.
4) Gabbasova Saule Telembaevna - RSE presso l'RSE "Istituto di ricerca kazako di oncologia e radiologia", capo del dipartimento di emoblastosi.
5) Karakulov Roman Karakulovich - Dottore in scienze mediche, professore, accademico del MAI RSE presso l'Istituto di ricerca kazako di oncologia e radiologia, ricercatore capo del dipartimento di emoblastosi.
6) Tabarov Adlet Berikbolovich - Capo del Dipartimento di Gestione Innovativa della RSE presso la RSE "Ospedale del Centro Medico Amministrazione del Presidente della Repubblica del Kazakistan" farmacologo clinico
Dichiarazione di assenza di conflitto di interessi:
Gli autori non hanno interessi finanziari concorrenti.
Revisori:
1) Afanasyev Boris Vladimirovich - Dottore in scienze mediche, direttore dell'Istituto di ricerca di oncologia, ematologia e trapianto infantile intitolato a R.M. Gorbacheva, capo del dipartimento di ematologia, transfusiologia e trapiantologia, istituto statale di bilancio per l'istruzione professionale superiore, prima università medica statale di San Pietroburgo. IP Pavlova.
2) Rakhimbekova Gulnar Ayapbekkyzy - Dottore in scienze mediche, professore, Centro medico scientifico nazionale JSC, capo del dipartimento.
3) Pivovarova Irina Alekseevna - Dottore in Medicina, Master in Business Administration, Capo ematologo freelance del Ministero della Sanità e dello Sviluppo Sociale della Repubblica del Kazakistan.
Indicazione delle condizioni per la revisione del protocollo: Il protocollo viene rivisto ogni 3 anni o quando diventano disponibili nuove prove.
Files allegati
Attenzione!
- Automedicando, puoi causare danni irreparabili alla tua salute.
- Le informazioni pubblicate sul sito web MedElement e nelle applicazioni mobili "MedElement", "Lekar Pro", "Dariger Pro", "Diseases: Therapist's Guide" non possono e non devono sostituire una consultazione faccia a faccia con un medico. Assicurati di contattare una struttura medica se hai malattie o sintomi che ti preoccupano.
- La scelta dei farmaci e il loro dosaggio devono essere discussi con uno specialista. Solo un medico può prescrivere il medicinale giusto e il suo dosaggio, tenendo conto della malattia e delle condizioni del corpo del paziente.
- Il sito web MedElement e le applicazioni mobili "MedElement", "Lekar Pro", "Dariger Pro", "Disseases: Therapist's Directory" sono esclusivamente informazioni e risorse di riferimento. Le informazioni pubblicate su questo sito non devono essere utilizzate per modificare senza autorizzazione le prescrizioni del medico.
- Gli editori di MedElement non sono responsabili per eventuali lesioni personali o danni alla proprietà derivanti dall'uso di questo sito.
La sindrome mielodisplastica è considerata una delle malattie gravi del sistema emopoietico. La malattia è difficile da trattare, il che non è sempre efficace. L'ulteriore prognosi dopo che la patologia è stata stabilita dipende da molte caratteristiche del decorso della malattia. Spesso solo la terapia radicale effettuata nelle prime fasi dello sviluppo della patologia può salvare la vita del paziente.
Idea generale della malattia
La sindrome MDS viene utilizzata dagli specialisti per designare uno spettro di varie patologie caratterizzate da citopenia nel sangue e dalla diffusione di alterazioni patologiche che colpiscono il midollo osseo.
Ogni malattia rappresenta un pericolo per l'uomo e può provocare lo sviluppo di una forma acuta di sindrome mieloblastica.
Gli esperti prestano sufficiente attenzione alla malattia, il che è dovuto all’aumento del numero di casi. Tuttavia, non esiste un regime terapeutico specifico.
Inoltre, la sindrome mielodisplastica inizia spesso a essere rilevata nei giovani. Secondo gli esperti ciò è dovuto alla cattiva situazione ambientale.
Il gruppo a rischio comprende principalmente persone di età superiore ai 50 anni. La patologia nei bambini si manifesta nei casi più estremi.
Nella medicina moderna, la sindrome mielodisplastica è divisa in forme primarie e secondarie. Il primo tipo viene spesso rilevato in pazienti di età superiore ai 60 anni. La sindrome secondaria viene stabilita indipendentemente dalla fascia di età ed è una complicazione di un'altra malattia.
Classificazione
La sindrome mielodisplastica è un gruppo di malattie specifiche che hanno un meccanismo di sviluppo simile e altre caratteristiche. Secondo l’OMS esistono diverse classificazioni.
Anemia refrattaria
La malattia si manifesta con un numero insufficiente di globuli rossi nel sangue. Ma con l'anemia refrattaria, il livello delle piastrine e dei leucociti non cambia e rimane invariato.
Il principale metodo diagnostico è il test del plasma. Se necessario, possono essere prescritti altri metodi di esame.
Anemia refrattaria con sideroblasti ad anello
I risultati di un esame del sangue rivelano un numero insufficiente di globuli rossi. Nelle cellule c'è un alto contenuto di ferro.
Ma nonostante i disturbi, il livello delle piastrine non cambia e rimane allo stesso livello.
Anemia refrattaria con mancanza di blasti
Con la malattia nel sangue c'è un numero insufficiente di blasti che sono nella fase di trasformazione.
Inoltre, nel plasma viene rilevata una mancanza di globuli rossi. Anche i livelli di piastrine e leucociti non rientrano nei limiti normali. Tuttavia le violazioni sono minori.
Il midollo osseo contiene non più del 19%, ma non meno del 5% di blasti.
Citopenia refrattaria
Molto spesso combinato con displasia multilineare. C'è una diminuzione di due o più indicatori nel sangue.
Non si osserva più del 5% dei blasti contenuti nel midollo osseo. Nel sangue che si muove lungo la periferia, il loro contenuto non supera l'1%.
Con il passare del tempo, in assenza di terapie o cure inadeguate, la patologia può trasformarsi in leucemia.
Sindrome MDS con una violazione del numero di cromosomi
C'è una mancanza di globuli rossi nel sangue. Il numero di blasti nel midollo osseo, come nella citopenia di tipo refrattario, non supera il 5%. Nel plasma non si trova più dell'1%.
I cromosomi subiscono alcuni cambiamenti, la cui causa è un processo patologico.
Nella letteratura medica si distingue la sindrome mielodisplastica di tipo non classificato. È caratterizzato da una significativa diminuzione della conta delle cellule del sangue. Allo stesso tempo, il livello dei blasti sia nel plasma che nel midollo osseo rimane in quantità normale e non subisce cambiamenti.
Quadro clinico
La sindrome mielodisplastica si manifesta sotto forma di debolezza e mancanza di respiro nelle fasi iniziali del suo sviluppo. Ma in alcuni casi la malattia può essere asintomatica.
Molto spesso, la patologia viene determinata in modo casuale quando un paziente dona il sangue per l'analisi in presenza di un'altra malattia o ai fini di un esame preventivo.
Nel corso del tempo si sviluppano sintomi che vengono spesso confusi con malattie del fegato o disturbi autoimmuni.
Il paziente lamenta i seguenti sintomi:
- Pallore pelle.
- Frequente raffreddori e ARVI.
- L'aspetto di punti sottocutanei emorragie.
Dopo lievi ferite e contusioni, si verifica un livido o un livido nel punto dell'impatto. Nel corso del tempo, quando la malattia progredisce e il paziente non riceve cure, altri segni si aggiungono ai sintomi principali.
Le manifestazioni cliniche comprendono lividi sottocutanei, che colpiscono una parte significativa della pelle, dolore alle articolazioni e alle ossa e perdita di peso.
Secondo i risultati di un esame del sangue clinico, si nota una diminuzione netta e significativa dell'emoglobina. I pazienti hanno difficoltà a respirare, ma non vi è alcun segno di asma.
Dopo uno sforzo fisico minore, appare debolezza e il corpo si stanca rapidamente. La perdita di peso corporeo avviene in un contesto di perdita di appetito. In alcuni casi, la temperatura corporea può salire fino a 40 gradi.
Tutti i sintomi hanno vari gradi di gravità, a seconda del periodo di sviluppo della malattia e delle caratteristiche del corpo del paziente.
Perché si verifica la sindrome mielodisplastica?
Gli scienziati non sono stati in grado di scoprire le vere ragioni dello sviluppo della patologia nemmeno dopo la ricerca. Ma sono stati identificati numerosi fattori che possono influenzare la composizione del sangue e provocare lo sviluppo della sindrome.
È stato dimostrato che il tipo primario di patologia colpisce più spesso le persone di età superiore ai 60 anni. La sua comparsa può essere influenzata da fattori quali predisposizione genetica, condizioni ambientali sfavorevoli, alti livelli di radiazioni e lavoro in condizioni di lavoro pericolose.
La MDS può verificarsi anche quando si lavora costantemente con sostanze tossiche, chimiche e velenose, a causa del fumo e del consumo di alcol. Gli esperti hanno scoperto che malattie ereditarie come la sindrome di Down, la neurofibromatosi e l'anemia di Fanconi hanno un impatto significativo sullo sviluppo della patologia.
La sindrome mielodisplastica secondaria, secondo i risultati degli studi, si verifica durante la chemioterapia, assumendo una serie di potenti farmaci o radioterapia.
Le MDS secondarie si verificano indipendentemente dalla fascia di età. Quando viene rilevata, la prognosi è spesso sfavorevole, poiché la malattia ha un decorso rapido e ha un impatto negativo su tutti gli organi e sistemi.
Metodi diagnostici
La sindrome mielodisplastica viene diagnosticata sulla base dei risultati degli esami del sangue di laboratorio e dell'istologia del midollo osseo. Il medico esamina anche l’anamnesi, che aiuta a determinare lo stile di vita del paziente, la presenza di rischi professionali e la predisposizione genetica.
Vengono inoltre prescritti numerosi metodi strumentali e test di laboratorio per stabilire un quadro completo della malattia.
Emogramma
Un metodo di analisi del sangue di laboratorio che rileva la presenza di anemia, neutropenia o monocitosi. Quando viene stabilita la pencitopenia, al paziente viene prescritto un esame istologico di campioni di midollo osseo.
Analisi biochimica
Aiuta a determinare il livello di ferro nel sangue, nonché di acido folico, urea e fosfatasi alcalina.
Il sangue deve essere donato al mattino a stomaco vuoto. Prima della procedura è vietato fare esercizio fisico, mangiare, stressarsi e fumare.
Immunogramma
Questo è un esame del sangue completo che aiuta uno specialista a determinare lo stato di immunità.
Gli indicatori ridotti indicano che le difese sono soppresse e il corpo non è in grado di far fronte da solo alla malattia.
Istologia
Prima degli esami di laboratorio viene eseguita una biopsia in cui uno specialista preleva un campione di midollo osseo. Per questo viene utilizzato un dispositivo speciale, ad un'estremità del quale è presente un ago speciale.
I campioni risultanti vengono inviati al laboratorio, dove vengono esaminati al microscopio. Sulla base dei risultati dello studio, viene determinata la presenza o l'assenza di cellule tumorali.
Studio citochimico
Questo metodo diagnostico consente di identificare disturbi nel metabolismo delle vitamine e di vari microelementi nel corpo.
Ecografia, TC e RM
L'ecografia, la tomografia computerizzata o la risonanza magnetica aiutano a determinare la disfunzione degli organi interni.
Sulla base della ricerca ottenuta, lo specialista determina il grado di sviluppo della malattia, il grado di cambiamento nel funzionamento degli organi interni, la diminuzione dell'immunità e altre caratteristiche del decorso della malattia.
Inoltre, il medico deve effettuare una diagnosi differenziale, poiché la sindrome mielodisplastica è simile nelle sue manifestazioni cliniche alle seguenti malattie:
- Leucemia forma acuta.
- Malattie fegato.
- Disturbo metabolico proteina composti nel corpo.
- Avvelenamento tossico e sostanze e fumi tossici.
- Linfoma di natura maligna.
- Mielosoppressivo sindrome.
Solo dopo uno studio completo e uno studio dei risultati uno specialista può formulare una diagnosi accurata ed eseguire la terapia.
Trattamento della sindrome MDS
Si ritiene che l'unico trattamento efficace per stabilire la sindrome MDS sia un trapianto di midollo osseo. Ma questo metodo, prima di tutto, non porta sempre al risultato desiderato e presenta una serie di svantaggi: il costo della procedura è piuttosto elevato, la probabilità di rigetto delle cellule trapiantate è elevata e la necessità di condurre una serie di studi per preparare il paziente al trapianto.
Spesso l'operazione può essere rinviata a tempo indeterminato a causa dell'assenza di un donatore.
Prima della procedura di trapianto di cellule del midollo osseo, il paziente dovrà sottoporsi a un ciclo di chemioterapia. Ma il metodo non porta sempre il risultato desiderato. Inoltre, dopo un ciclo di trattamento, si verificano effetti collaterali sotto forma di danni alle cellule degli organi interni, perdita di capelli, perdita di unghie, nausea e una significativa diminuzione dell'immunità.
Per la chemioterapia vengono utilizzati farmaci moderni come la decitabina o la citarabina. Il dosaggio dei farmaci viene selezionato individualmente in base al grado di sviluppo della patologia, alle condizioni e all'età del paziente, nonché ad altre caratteristiche del decorso della malattia.
Oggi gli esperti ritengono anche che il trapianto di cellule staminali sia un modo efficace per curare la patologia. Ma dopo la procedura potrebbero verificarsi effetti indesiderati. Il trapianto di cellule staminali può ridurre il rischio di leucemia nella fase acuta, che mette a rischio la vita del paziente.
Nelle fasi iniziali della malattia viene utilizzata una trasfusione di sangue, grazie alla quale i sintomi spiacevoli scompaiono completamente. Il sangue del donatore viene spesso somministrato sotto forma di globuli rossi o concentrato di piastrine.
In alcuni casi viene prescritta una terapia di accompagnamento, che viene eseguita in un breve periodo di tempo. Questo perché livelli eccessivi di ferro nel sangue possono causare gravi complicazioni.
Per stimolare la produzione di cellule del sangue vengono prescritti farmaci come Leukin, Neupogen o Eritropoietina.
Quando viene diagnosticata la sindrome mielodisplastica, al paziente vengono prescritti farmaci per aumentare e mantenere le difese dell'organismo. Per escludere l'insorgenza della leucemia, vengono solitamente somministrati per via endovenosa.
Possibili complicazioni
La sindrome mielodisplastica è caratterizzata da danni al midollo osseo e cambiamenti nel numero delle cellule del sangue. Senza trattamento, si sviluppa l’anemia.
Sullo sfondo di cellule del sangue insufficienti, si osserva lo sviluppo di insufficienza cardiaca e aumenta significativamente la probabilità di infezione da virus, funghi e microrganismi infettivi.
La sindrome MDS, complicata dall'anemia, è caratterizzata da affaticamento. Vertigini frequenti. Tuttavia, si ritiene che la malattia sia lieve.
A causa dell'insufficienza delle cellule del sangue, l'immunità diminuisce e il corpo diventa suscettibile a varie malattie infettive. Ciò porta allo sviluppo di polmonite, stomatite, laringite e altre patologie.
Quando il numero di piastrine è insufficiente si verifica un disturbo della coagulazione del sangue. Per questo motivo, anche lievi lesioni ai tessuti molli possono essere fatali.
Cancro del sangue e sindrome MDS
La sindrome MDS e il cancro del sangue hanno una stretta relazione. Uno stadio avanzato della malattia porta spesso alla trasformazione di cellule sane.
Ma la comparsa del cancro non si verifica in tutti i casi.
Il cancro del sangue viene diagnosticato solo quando la sindrome mielodisplastica è una malattia secondaria. Le mutazioni cellulari sono causate dai principi attivi dei farmaci utilizzati per la chemioterapia. In questo caso, la malattia si presenta in forma acuta e quasi non è suscettibile al trattamento farmacologico.
Quanto vivono?
L'ulteriore prognosi una volta stabilita la sindrome dipende da molti fattori. Il tipo patogenetico della patologia è di notevole importanza.
Una prognosi più favorevole si osserva quando la malattia è di tipo primario. Con l'aiuto della terapia radicale è possibile aumentare significativamente l'aspettativa di vita del paziente.
Una prognosi sfavorevole viene stabilita nei casi in cui la sindrome ha iniziato a svilupparsi durante l'assunzione di farmaci chemioterapici e ha una forma secondaria. In questo caso, molto spesso si trasforma in cancro del sangue.
In media, se viene rilevato un elevato grado di rischio, l’aspettativa di vita non supera i sei mesi.
Questo è il motivo per cui dovresti consultare un medico se si verificano sintomi. I segni non sempre indicano questa malattia, ma una diagnosi tempestiva aiuterà a ridurre significativamente il rischio di complicanze.
Prevenzione
Non esistono regole speciali per prevenire lo sviluppo della sindrome mielodisplastica. Gli esperti raccomandano di utilizzare misure generali.
Per ridurre il rischio di sviluppare tumori del sangue, si consiglia ai pazienti di rispettare i seguenti requisiti:
- Rafforzare immunità. Per fare questo, devi praticare sport e indurire te stesso. I multivitaminici aiuteranno a sostenere le difese del corpo.
- Giusto mangiare. La dieta dovrebbe includere frutta, verdura e bacche. Devi rinunciare al fast food e al fast food.
- Mantenere il livello emoglobina al giusto livello. Puoi scoprire gli indicatori facendo un esame del sangue per l'emoglobina. I risultati possono essere ottenuti dal medico dopo 2-7 giorni.
- Quotidiano camminare all'aria fresca. Anche una passeggiata di cinque minuti ti gioverà. Ma prima di uscire, dovresti assolutamente vestirti per il tempo, per non congelare e sudare.
- La pelle deve essere protetta dall'esposizione chimico sostanze.
I pazienti dovrebbero sottoporsi tempestivamente agli esami del sangue e visitare regolarmente il proprio medico per esami preventivi.
La sindrome mielodisplastica si riferisce a gravi lesioni del midollo osseo, caratterizzate da cambiamenti nella composizione del sangue. È importante effettuare il trattamento in modo tempestivo, poiché la malattia può causare gravi conseguenze.
Un insieme di malattie caratterizzate da una diminuzione di uno o più tipi di cellule nel sangue periferico (citopenia) e dall’interruzione dello sviluppo delle cellule progenitrici nel midollo osseo è chiamata “sindrome mielodisplastica” (MDS). L'anemia refrattaria è inclusa nel gruppo di queste malattie e la sua presenza può provocare leucemia acuta.
La sindrome mielodisplastica si manifesta nelle persone di età superiore ai sessant'anni, ma può manifestarsi anche nei trentenni.
Ragioni per la MDS
Non è stato ancora stabilito quale sia il fattore principale nella comparsa della sindrome mielodisplastica. Tuttavia, è generalmente accettato che le MDS possano essere primarie e secondarie. Il tipo primario si verifica spontaneamente e rappresenta l'80-90% di tutti i casi di patologia, mentre il tipo secondario è meno comune e, di regola, è una complicanza della chemioterapia o una conseguenza della predisposizione ereditaria.
I fattori che contribuiscono a influenzare lo sviluppo della malattia sono:
- avvelenamento cronico con benzina, pesticidi, solventi organici;
- età anziana e senile;
- l'effetto delle radiazioni ionizzanti;
- malattie del sangue geneticamente determinate.
Cosa succede durante la MDS
I processi fisiologici del midollo osseo sono accompagnati dalla produzione di cellule staminali, dalle quali si sviluppano i necessari componenti del sangue. La displasia del midollo osseo porta all'interruzione della normale formazione delle cellule del sangue: globuli rossi, globuli bianchi e piastrine. Come risultato di questa patologia, il processo di maturazione delle cellule staminali viene interrotto, motivo per cui le cellule immature che non sono in grado di far fronte alle loro funzioni entrano nel flusso sanguigno.
Tipi di sindrome mielodisplastica
Secondo la classificazione adottata dall’Organizzazione Mondiale della Sanità si distinguono i seguenti sottotipi di MDS:
- anemia refrattaria;
- anemia refrattaria con sideroblasti ad anello;
- anemia refrattaria con eccesso di blasti;
- anemia refrattaria con eccesso di blasti in trasformazione;
Segni di anemia refrattaria
I sintomi comuni di tutti i sottotipi di MDS comprendono debolezza, aumento dell'affaticamento, pallore della pelle e delle mucose, aumento della temperatura corporea, tendenza al sanguinamento e all'emorragia, nonché ingrossamento del fegato e della milza.
Anemia Come trattare l'anemia?
Anemia-sintomi e trattamento
9 POSSIBILI SEGNI DI ANEMIA NON NOTEVOLI A UN PRIMO SGUARDO
Anemia da carenza di ferro 1
Anemia.Sintomi.Cause.Trattamento
Cause dell'anemia - Dr. Komarovsky
Anemia da carenza di ferro | Cosa fare | Come trattare | Sintomi | gravidanza | Malattia | Dottor Phil
Le cose più importanti: anemia, nodo alla gola, vene varicose sul viso
L'anemia o l'anemia possono essere curate con rimedi popolari
Vegetarianismo/Anemia/Morte lenta
Anemia. Sintomi e tipi di anemia
Perché l’anemia è così spaventosa?
Anemia, trattamento
Le cose più importanti: anemia, spesso mal di stomaco, secchezza delle fauci
Tutto sul sangue. Anemia. Emoglobina. Olga Butakova ACCADEMIA DELLA SALUTE
Alimentazione per l'anemia
ANEMIA. COME TRATTARE. SINTOMI. ANALISI. FGS. CONTAGOCCE.COMPRESSE. PERDITA DEI CAPELLI #anemia
B 12 - ANEMIA DA CARENZA
Anemia. Come aumentare l'emoglobina con mezzi naturali?
Torsunov O.G. Sulle cause dell'anemia da carenza di ferro
L'anemia refrattale è caratterizzata dall'assenza di effetto terapeutico quando si consumano preparati di ferro e vitamine. Un esame del sangue mostrerà una leggera displasia di una sola classe di cellule del sangue e nell'aspirato di midollo osseo il numero di blasti sarà inferiore al 5%.
Nell'anemia refrattaria con sideroblasti, si verificheranno disturbi solo nella serie emopoiesi dei globuli rossi, nonché la presenza di oltre il 15% di sideroblasti (globuli rossi anormali con inclusione di granuli di ferro). Questo tipo di anemia è considerato il più favorevole.
L'anemia refrattaria con eccesso di blasti è divisa in due sottogruppi, i cui criteri sono determinati dal numero di blasti nel midollo osseo. Nei pazienti del primo gruppo il contenuto di blasti è del 5-9%, nel secondo gruppo del 10-19%.
L'anemia refrattaria con eccesso di blasti in trasformazione è caratterizzata dal 5% di queste cellule nel sangue periferico e dal 20-30% nell'aspirato midollare. Inoltre, i bastoncini di Auer vengono rilevati nelle cellule precursori dei granulociti.
Diagnosi di MDS
La diagnosi viene chiarita se vi sono segni di anemia, nonché con l'aiuto di esami del sangue di laboratorio, puntura del midollo osseo ai fini dell'analisi istologica e citologica, nonché con la determinazione delle patologie cromosomiche nel sangue periferico mediante ricerca citogenetica.
Trattamento della sindrome mielodisplastica
L’unico modo per risolvere il problema della sindrome mielodisplastica è il trapianto di midollo osseo. Altri metodi mirano a controllare i sintomi, prevenire complicanze e migliorare la qualità della vita dei pazienti. La scelta dello schema di intensità del trattamento viene effettuata tenendo conto dell’età del paziente, nonché confrontando rischi e benefici. Il trattamento dei pazienti con MDS viene effettuato da un oncologo e da un ematologo. L’inizio precoce del trattamento aumenta la possibilità di una remissione riuscita della sindrome mielodisplastica.
La terapia a bassa intensità ha lo scopo di ridurre i segni di anemia e comprende la trasfusione di globuli rossi, piastrine e la somministrazione di eritropoietine. Gli agenti chemioterapici vengono utilizzati anche in dosi minime (citarabina, decitarabina).
Il trattamento ad alta intensità è coerente con i regimi chemioterapici per la leucemia mieloblastica acuta.
Sindrome mielodisplastica: prognosi della malattia
I dati prognostici per la sindrome mielodisplastica saranno determinati direttamente dal tipo di patologia, dall'età del paziente e dalle malattie concomitanti.
La sindrome mielodisplastica è un gruppo di malattie del sangue clonale eterogenee accomunate dalle seguenti caratteristiche: emopoiesi inefficace, citopenia periferica, displasia in uno o più linee emopoietiche con un alto potenziale di trasformazione in leucemia mieloide acuta.
L'ematopoiesi insufficiente si manifesta con anemia, aumento del sanguinamento e suscettibilità alle infezioni. La sindrome mielodisplastica (MDS) si manifesta in persone di qualsiasi età, compresi i bambini, ma le persone di età superiore ai 60 anni sono più suscettibili ad essa.
Secondo l'ICD-10, alle sindromi mielodisplastiche viene assegnato il codice D46.
Cause
Le cellule del sangue vengono sintetizzate e maturano principalmente nel midollo osseo (questo processo è chiamato mielopoiesi e il tessuto in cui avviene è chiamato mieloide), quindi, dopo aver adempiuto alla loro funzione e invecchiato, vengono distrutti dalla milza e ne prendono di nuove il loro posto. Con la sindrome mielodisplastica, il midollo osseo perde la capacità di riprodurre le cellule del sangue (tutti - globuli rossi, leucociti, piastrine o solo alcune) nella quantità richiesta dall'organismo, le cellule immature (blasti) entrano nel sangue, a seguito della quale svolge peggio le sue funzioni. Ciò si manifesta con sintomi caratteristici della MDS. In circa il 30% dei casi, il processo di mielopoiesi diventa completamente incontrollato nel tempo, il numero di forme esplosive delle cellule del sangue aumenta, sostituendo le cellule normali e mature. Quando il numero di blasti nel sangue supera il 20% (prima la soglia era del 30%), viene posta la diagnosi di leucemia mieloide acuta.
A seconda che la causa della disfunzione del midollo osseo sia nota o meno, le MDS vengono suddivise in primarie, o idiopatiche, e secondarie. Secondaria si verifica a seguito dell'inibizione della funzione del midollo osseo dopo la chemioterapia o l'esposizione alle radiazioni. Tale effetto rientra solitamente nella terapia antitumorale, cioè effettuata per alcuni tipi di cancro. In questo caso, la MDS può essere considerata una complicazione.
La MDS primaria o idiopatica si verifica spontaneamente, senza alcuna patologia precedente e per una ragione sconosciuta. È possibile che il fattore predisponente sia genetico, poiché in alcuni tipi di sindrome si riscontrano alterazioni cromosomiche.
I fattori che contribuiscono allo sviluppo della MDS sono:
- fumare;
- contatto con sostanze chimiche cancerogene (pesticidi, erbicidi, benzene);
- esposizione a radiazioni ionizzanti;
- età anziana.
Forme della malattia
Come accennato in precedenza, le MDS si dividono in due tipologie, primarie e secondarie.
Le MDS primarie sono più comuni (circa l'80% di tutti i casi), la maggior parte dei pazienti sono anziani (65-75 anni). Anche le MDS secondarie colpiscono soprattutto gli anziani, poiché in loro sono più frequenti i tumori maligni e quindi le loro complicanze. Le MDS secondarie rispondono meno alla terapia ed sono associate a una prognosi peggiore.
Inoltre, le MDS sono suddivise in tipologie cliniche a seconda del tipo di blasti, del loro numero e della presenza di alterazioni cromosomiche, questa classificazione è proposta dall'Organizzazione Mondiale della Sanità (OMS). Secondo la classificazione dell’OMS si distinguono le seguenti forme di MDS:
- anemia refrattaria (cioè resistente alla terapia classica);
- citopenia refrattaria con displasia multilineare;
- MDS con delezione isolata 5q;
- MDS non classificabile;
- anemia refrattaria con sideroblasti a forma di anello;
- Citopenia refrattaria con displasia multilineare e sideroblasti ad anello;
- anemia refrattaria con eccesso di blasti-1;
- anemia refrattaria con eccesso di blasti-2.
Fasi della malattia
Nel corso della MDS si distinguono tre fasi, che però non sono sempre clinicamente chiaramente distinguibili l'una dall'altra; le differenze vengono determinate in laboratorio. Questi sono lo stadio dell'anemia, lo stadio di trasformazione (intermedio tra l'anemia e la leucemia acuta) e la leucemia mieloide acuta. Non tutti i ricercatori concordano con la definizione di leucemia mieloide acuta come stadio della sindrome mielodisplastica, poiché si tratta di una malattia mieloproliferativa (cioè caratterizzata da una crescita cellulare incontrollata), che quindi non soddisfa pienamente le caratteristiche della MDS.
Sintomi
I principali sintomi delle MDS sono associati a manifestazioni di anemia. I pazienti lamentano un aumento dell'affaticamento, attacchi di vertigini, mancanza di respiro durante l'attività fisica, che in precedenza era facilmente tollerata. L’anemia è associata ad una ridotta produzione di globuli rossi, con conseguente bassi livelli di emoglobina nel sangue.
In alcuni casi si sviluppa la sindrome emorragica, caratterizzata da un aumento del sanguinamento. Il paziente inizia a notare che anche lesioni superficiali minori causano sanguinamento che non si arresta per lungo tempo, possono comparire gengive sanguinanti, sangue dal naso frequente e spontaneo, petecchie sulla pelle e sulle mucose, nonché ematomi multipli (lividi), sia senza connessione con qualsiasi trauma ricordato dal paziente, o dopo un piccolo livido o addirittura pressione. La sindrome emorragica è associata a disturbi della trombocitopoiesi.
I pazienti affetti da MDS sono anche suscettibili alle malattie infettive. Soffrono spesso di raffreddori, infezioni batteriche e fungine della pelle. Questa condizione è causata dalla neutropenia (carenza di neutrofili).
Inoltre, i segni di MDS possono includere:
- aumento senza causa della temperatura, spesso a valori elevati (38 °C e oltre);
- perdita di peso, diminuzione dell'appetito;
- epatomegalia;
- splenomegalia;
- sindrome del dolore.
In alcuni casi la MDS non si manifesta in alcun modo e viene scoperta per caso durante un esame del sangue di laboratorio per un altro motivo.
Diagnostica
Il metodo principale per diagnosticare la MDS è il laboratorio. Se si sospetta mielodisplasia, si esegue quanto segue:
- Esame del sangue clinico. In questo caso vengono rilevati anemia (macrocitica), reticolocitopenia, leucopenia, neutropenia e con la sindrome 5q - trombocitosi. Circa la metà dei pazienti presenta pancitopenia.
- Biopsia del midollo osseo. La citosi è solitamente normale o aumentata, ma in circa il 10% dei pazienti è ridotta (variante ipoplastica della MDS), vi sono segni di alterazione dell'emopoiesi di una o più emopoiesi e un aumento del contenuto di forme blastiche e sideroblasti patologici (eritrociti contenenti ferro depositi) possono essere rilevati. Per identificare i fenotipi anomali viene studiato l'immunofenotipo delle cellule del midollo osseo; ciò consente la diagnosi differenziale delle MDS e delle citopenie non clonali, importante per la prognosi.
- Analisi citogenetica. Anomalie citogenetiche clonali si riscontrano nel 40-70% dei pazienti, in particolare la delezione (monosomia) del cromosoma 7 (7q), che è prognosticamente sfavorevole.
- Determinazione dei livelli sierici di ferro e feritina. I livelli sono stati aumentati.
- Determinazione dell'eriropoietina endogena (con<500 МЕ/л эритропоэз-стимулирующие агенты обычно обеспечивают хороший терапевтический ответ).
Nel 95% dei casi la diagnosi viene posta sulla base dell'analisi citologica e istologica del midollo osseo.

Criteri diagnostici
Per determinare la MDS sono stati sviluppati criteri speciali, ovvero le condizioni in cui viene posta questa diagnosi. I criteri diagnostici sono i seguenti:
- Citopenia periferica del lignaggio 1, 2 o 3 (cioè rilevata nel sangue periferico);
- displasia: segni di alterata emopoiesi in almeno il 10% delle cellule in almeno una linea emopoietica;
- cambiamenti citogenetici caratteristici (presenza di un clone patologico).
La citopenia deve essere stabile e osservata per almeno sei mesi, ma se viene rilevato un cariotipo specifico, o è accompagnata da displasia di almeno due emopoiesi, sono sufficienti due mesi.
Per fare una diagnosi devono essere escluse altre malattie accompagnate da displasia cellulare e citopenia.
Se la citopenia viene rilevata senza altri segni di MDS, viene diagnosticata citopenia idiopatica, il cui significato non è stato stabilito; quando viene rilevata displasia senza citopenia - displasia idiopatica, il cui significato non è stato stabilito. Ciò richiede un monitoraggio costante del paziente con un esame del midollo osseo ripetuto dopo 6 mesi, poiché entrambe queste diagnosi possono progredire in MDS e leucemia mieloide acuta (o altra malattia mieloproliferativa).
Diagnosi differenziale
Le MDS si differenziano dalle seguenti malattie:
- anemia (principalmente megaloblastica, sideroblastica e aplastica);
- leucemia mieloide acuta;
- leucopenia con neutropenia;
- trombocitopenia immunitaria primaria;
- emopoiesi clonale di potenziale incerto;
- mielofibrosi primaria;
- grave intossicazione di varie eziologie.
Trattamento
Nel 1997 è stata sviluppata una scala speciale, chiamata scala IPSS (International Scoring Prognostic System), dividendo i pazienti in gruppi a rischio. In base a un determinato gruppo di rischio, vengono selezionate le tattiche terapeutiche e, come suggerisce il nome, viene valutata la prognosi.
I punti vengono assegnati in base a tre fattori:
- numero di forme esplosive;
- il numero di germogli ematopoietici colpiti;
- categoria citogenetica.
La somma dei punti consente di classificare il paziente in uno o in un altro gruppo a rischio:
|
Conversione in leucemia mieloide acuta nel 23% dei pazienti (anni) |
Sopravvivenza mediana (anni) |
% dei pazienti |
||
|
Intermedio 1 |
||||
|
Intermedio 2 |
||||
Il metodo di trattamento dipende dalla categoria di rischio, dalle condizioni e dall'età del paziente. In caso di MDS asintomatica, ai pazienti appartenenti al gruppo a rischio basso o intermedio potrebbe non essere prescritta la terapia; è richiesta solo l'osservazione dinamica.
Trapianto allogenico di cellule staminali emopoietiche
Questo è l'unico metodo radicale, cioè che consente di ottenere il recupero, per trattare le MDS. È indicato per i pazienti classificati a rischio intermedio e alto 2 e a rischio intermedio 1 con elevate percentuali di blasti o caratteristiche citogenetiche sfavorevoli. L'età dei pazienti è prevalentemente fino a 60 anni (questo criterio è in fase di revisione a causa del miglioramento del metodo; anche i pazienti più anziani vengono considerati candidati al trapianto). Il trapianto allogenico richiede la presenza di un donatore identico.

Altri trattamenti
Oltre al trapianto di cellule staminali, possono essere utilizzati:
- Terapia intensiva di induzione. Indicato per pazienti di età inferiore a 70 anni appartenenti ad un gruppo ad alto rischio senza alterazioni citogenetiche sfavorevoli in buone condizioni funzionali senza patologia concomitante, con conta blastica ≥10%.
- Terapia con azatidina. Indicato per pazienti appartenenti a gruppi con 2 rischi intermedi e alti, non idonei al trapianto allogenico di cellule staminali emopoietiche, nonché pazienti con sintomi appartenenti a gruppi a rischio basso e 1 intermedio. Il trattamento viene continuato fino alla progressione della malattia o alla comparsa di tossicità.
- La terapia con lenalidomide è indicata per la sindrome 5q-.
- La terapia immunosoppressiva combinata (globulina antimonocitaria + ciclosporina) è indicata nei pazienti di età inferiore a 60 anni con cariotipo e contenuto blastico normali<5 %, коротким периодом зависимости от переливания эритроцитарной массы (менее 6 месяцев) и наличием HLA-DR15, или наличием клона пароксизмальной ночной гемоглобинурии.
- Trasfusione di globuli rossi e piastrine.
- Terapia con fattori di crescita emopoietici (eritropoietina ricombinante, EPO).
- Assunzione di farmaci immunosoppressori (solitamente globulina antitimocitaria + ciclosporina).
- Chemioterapia a basso dosaggio (solitamente decitabina o citarabina) – per pazienti a rischio intermedio e alto con controindicazioni alla chemioterapia ad alte dosi.
Vengono utilizzati anche altri regimi terapeutici.
Possibili complicazioni e conseguenze
La MDS è una grave malattia del sangue che si trasforma in leucemia mieloide acuta nel 30% dei pazienti.
Sindrome mielodisplastica: prognosi
La prognosi dipende dal gruppo a rischio a cui appartiene il paziente. I pazienti a basso rischio hanno una sopravvivenza media di 6 anni dopo la diagnosi. Nei pazienti ad alto rischio: 6 mesi o meno. Il trapianto allogenico di cellule staminali emopoietiche garantisce una sopravvivenza a cinque anni nel 40-50% dei pazienti. Il trattamento correttamente selezionato aiuta ad aumentare la sopravvivenza nei pazienti ad alto rischio fino a un anno.
video
Ti offriamo la visione di un video sull'argomento dell'articolo.
Gli anni '30 del XX secolo furono un periodo di rapido sviluppo della medicina e di speranze per il trionfo universale del progresso. Ma la vita è una donna dura e difficile da affrontare. In primo luogo, il mondo ha dovuto affrontare la guerra, e poi gli scienziati, che 20 anni fa ardevano di sogni, hanno detto all'umanità che il cancro è il flagello del nostro tempo. La sindrome mielodisplastica, di cui parleremo oggi, non è una di quelle comuni, ma trova regolarmente le sue vittime. Cosa fa più spesso un paziente quando riceve una diagnosi deludente? Esatto, inizia a piangere il suo destino e si scoraggia. E invece di combattere il problema, si arrende. Non è necessario parlare di quale sarà il finale in questo caso.
Perché abbiamo deciso di iniziare con un'introduzione così deludente? La risposta è semplice. Devi capire che all'attuale livello di sviluppo della medicina, l'oncologia è proprio una diagnosi (dove la prognosi è tutt'altro che ovvia), e non un motivo per redigere un testamento. Difficile, che richiede il massimo sforzo da parte del paziente e dei suoi parenti, e da parte dei medici: fiducia incondizionata nel buon esito del trattamento. In altre parole, l’oncologia moderna non è solo (e non tanto!) tecniche all’avanguardia, farmaci super efficaci e attrezzature costose, ma anche mentalità per il successo e fiducia in un piccolo, ma tanto desiderato e atteso miracolo. Per favore, ricordalo!
Bisogna conoscere il nemico di vista: capiamo la teoria
La sindrome mielodisplastica non è una patologia isolata, come erroneamente crede la gente comune, ma un gruppo di malattie che colpiscono il midollo osseo, responsabile della produzione di sangue. In una persona sana, la naturale perdita di cellule viene compensata, motivo per cui il loro livello rimane approssimativamente lo stesso. In altre parole, in questo caso c'è una sorta di circolazione sanguigna nel corpo: la milza la “distrugge” e il midollo osseo la produce. La sindrome sconvolge l’equilibrio stabilito, causando un abbassamento del livello delle piastrine, dei globuli rossi o dei globuli bianchi. La prognosi (a riguardo alla fine dell'articolo) è condizionatamente sfavorevole.
Pertanto, questa malattia è considerata in un certo senso “scomoda”: dopotutto, rimuovere un tumore in un organo (fegato, polmoni o stomaco) e “soffocare” le cellule tumorali rimanenti con chemioterapia o radiazioni è una cosa, ma il trattamento del midollo osseo è efficace e sicuro per il paziente - completamente diverso.
È anche importante ricordare che alcuni tipi di malattie (anemia refrattaria con eccesso di blasti) sono correlati al problema che stiamo considerando, ma si distinguono e richiedono un approccio diverso al trattamento, sebbene la loro diagnosi sia standard per tali patologie. Per questo motivo, il paziente è costretto a restare negli studi medici per settimane prima che i medici capiscano di quale problema hanno a che fare. Nel frattempo, l’anemia refrattaria distruggerà il corpo a tal punto che non potrà essere offerto altro trattamento oltre al trattamento palliativo.
Un altro punto che incide negativamente sulla prognosi di guarigione riguarda l’età. La sindrome nella stragrande maggioranza dei casi viene diagnosticata nelle persone anziane, coloro che, a causa della loro età, hanno “acquisito” un insieme di malattie e le risorse protettive del corpo sono quasi esaurite.
Classificazione e tipologie esistenti
 1. RA - anemia refrattaria
1. RA - anemia refrattaria
- alterazioni del sangue: anemia, assenza di esplosioni;
- alterazioni del midollo osseo: displasia della linea eritrocitaria, blasti< 5%, кольцевых сидеробластов < 15%.
2. RCMD - citopenia refrattaria (con displasia multilineare)
- alterazioni del sangue: citopenia (due linee)/pancitopenia, assenza di blasti e bastoncini di Auer, monociti< 1х10 9 ;
- < 5%, нет палочек Ауэра, кольцевых сидеробластов < 15%.
3. 5q - sindrome mielodisplastica (con delezione isolata 5q)
- alterazioni del sangue: anemia, esplosioni< 5%, тромбоциты в норме;
- alterazione del midollo osseo: blasti< 5%, нет палочек Ауэра, изолированная делеция (5q).
4. MDS-N - sindrome mielodisplastica non classificata
- alterazioni del sangue: citopenia, assenza di esplosioni e bastoncini di Auer;
- alterazioni del midollo osseo: displasia di linee singole (granulocitiche o megacariocitiche), blasti< 5%, нет палочек Ауэра.
5. RAX - anemia refrattaria (con sideroblasti a forma di anello)
- alterazioni del sangue: anemia, assenza di esplosioni;
- alterazioni del midollo osseo: displasia isolata di un ceppo (eritroite), blasti< 5%, кольцевых сидеробластов > 15%.
6. RCMD-KS - combinazione di 2 e 5 tipi
- alterazioni del sangue: citopenia, assenza di blasti e bastoncini di Auer, monociti< 1х10 9 ;
- alterazioni del midollo osseo: displasia estesa (più del 10%), blasti< 5%, нет палочек Ауэра, кольцевых сидеробластов > 15%.
7. RAEB-1 - anemia refrattaria caratterizzata da un eccesso di blasti-1
- alterazioni del sangue: citopenia, esplosioni< 5%, нет палочек Ауэра, моноцитов < 1х10 9 ;
- alterazioni del midollo osseo: displasia generale (uno o più germogli), blasti dal 5% al 9%.
8. RAEB-2 - anemia refrattaria, caratterizzata da un eccesso di blasti-2
- alterazioni del sangue: citopenia, blasti dal 5% al 19%, bastoncini di Auer, possono essere presenti monociti< 1х10 9 ;
- alterazioni del midollo osseo: displasia generale (uno o più germogli), blasti dal 10% al 19%, sono presenti bastoncini di Auer.
Gold standard per la diagnosi

La diagnostica è una scienza molto imprecisa. E, sebbene il complesso di tutti i metodi di cui sopra sia considerato oggi completo e sufficiente, a causa del fattore età (praticamente non ci sono bambini con la sindrome, la maggior parte dei pazienti ha già superato la soglia dei 60 anni), la sua implementazione è irta di notevoli difficoltà. E il punto qui non è tanto l'elevato carattere traumatico delle procedure, ma piuttosto i cambiamenti naturali legati all'età. Pertanto, fare una diagnosi corretta è un compito non banale e piuttosto complesso.
Alcune malattie e patologie (l'elenco è riportato di seguito) possono presentare sintomi simili, quindi è necessario ricordare che la diagnosi deve essere differenziale:
- varie emoblastosi (eritremia, LMA - leucemia mieloblastica acuta, policitemia vera);
- linfomi maligni di varie eziologie;
- alcune malattie autoimmuni (la prognosi stessa è sfavorevole);
- danno tossico al corpo;
- sindrome mielodepressiva;
- HIV (se il trattamento con farmaci antiretrovirali altamente attivi non è stato effettuato);
- disturbi del metabolismo proteico;
- malattie epatiche croniche;
- glicogenosi.
Reclami e manifestazioni cliniche

Possibili fattori di rischio
- disturbi genetici o cromosomici;
- contatto prolungato con sostanze chimiche dannose senza protezione adeguata;
- esposizione alle radiazioni.
L'elenco sopra è molto condizionato e viene spiegato in modo abbastanza semplice. La sindrome è una di quelle malattie le cui cause profonde non sono state ancora completamente identificate e le teorie esistenti, con un approccio imparziale, non resistono alle critiche e possono essere confutate.
Principi del trattamento moderno

Se tutti i metodi elencati sono inefficaci per un motivo o per un altro (età, stadio della malattia, patologie concomitanti, sintomi gravi), può essere utilizzato un trattamento palliativo. Non è in grado di aiutare il paziente a riprendersi, ma può alleviare il dolore e migliorare la qualità della vita. Notiamo in particolare: proprio la vita (non importa quanto breve possa rivelarsi), e non la vegetazione senza scopo, sola con i propri problemi.
Previsione
Se ci concentriamo sul sistema prognostico WPSS sviluppato dall’OMS, l’efficacia del trattamento dipende da tre fattori principali:
- cariotipo (cattivo, mediocre, buono): da 2 a 0 punti;
- tipo di malattia: RAEB-2 - 3 punti; RAEB-1 - 2 punti; RCMD, RCMD-KS - 1 punto; RA, 5q, RAKS - 0 punti;
- necessità di trasfusioni di sangue: sì - 1 punto, no - 0 punti.
Tutti i punti vengono riassunti e sulla base di essi viene derivato un indicatore del gruppo di rischio, che fornisce un'idea approssimativa della possibile durata della vita:
- 0 punti: 136 mesi;
- 1 punto: 63 mesi;
- 2 punti: 44 mesi;
- 3-4 punti: 19 mesi;
- 5-6 punti: 8 mesi.
Ma qui ci sono due cose da ricordare. Primo: la medicina non si ferma, quindi è possibile che tra qualche anno la situazione cambi in modo significativo in meglio. Secondo: questi sono dati medi, statisticamente medi, e fare affidamento su di essi non è la decisione giusta. Ancora una volta, la sindrome mielodisplastica è una malattia il cui trattamento dipende in gran parte dalla fiducia del paziente in un risultato migliore.
- In contatto con 0
- Google+ 0
- OK 0
- Facebook 0






