Quando si studia la natura dell'eredità di vari tratti negli esseri umani, vengono descritti tutti i tipi conosciuti di ereditarietà e tutti i tipi di dominanza. Molti tratti vengono ereditati monogenico, cioè. sono determinati da un gene e vengono ereditati secondo le leggi di Mendel. Sono stati descritti più di mille tratti monogenici. Tra questi ci sono sia quelli autosomici che quelli legati al sesso. Alcuni di essi sono elencati di seguito.
Le malattie monogeniche si verificano nell'1-2% della popolazione mondiale. Questo è molto. La frequenza delle malattie monogeniche sporadiche riflette la frequenza del processo mutazionale spontaneo. Tra queste, gran parte sono malattie con un difetto biochimico. Un tipico esempio è fenilchetonuria.
manifestazione familiare
La sindrome di Morfan
Si tratta di una grave malattia ereditaria causata da una mutazione di un singolo gene che interrompe il normale ciclo di conversione della fenilalanina. Nei pazienti, questo amminoacido si accumula nelle cellule. La malattia è accompagnata da gravi sintomi neurologici (irritabilità), microcefalia (testa piccola) e alla fine porta all'idiozia. La diagnosi viene fatta biochimicamente. Attualmente, lo screening del 100% dei neonati per la fenilchetonuria viene effettuato negli ospedali di maternità. La malattia è curabile se il bambino viene trasferito in tempo a una dieta speciale che esclude la fenilalanina.
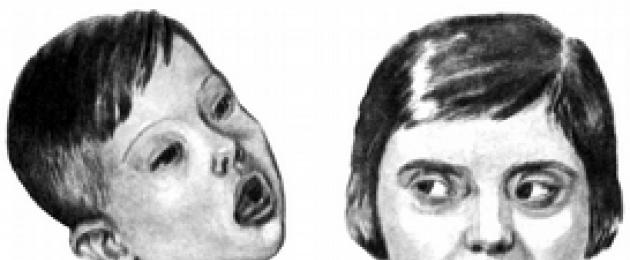
Un altro esempio di malattia monogenica è Sindrome di Morfan o malattia delle dita di ragno. Una mutazione dominante in un gene ha un forte effetto pleiotropico. Oltre all'aumento della crescita degli arti (dita), i pazienti presentano astenia, malattie cardiache, dislocazione del cristallino e altre anomalie. La malattia procede sullo sfondo di una maggiore intelligenza, in relazione alla quale è chiamata la "malattia delle grandi persone". Era malato, in particolare, il presidente americano A. Lincoln e l'eccezionale violinista N. Paganini.
Molte malattie ereditarie sono associate a un cambiamento nella struttura dei cromosomi o nel loro numero normale, ad es. con mutazioni cromosomiche o genomiche. Pertanto, una grave malattia ereditaria nei neonati, nota come “ sindrome del gatto che piange”, è causato dalla perdita (delezione) del braccio lungo del 5° cromosoma. Questa mutazione provoca uno sviluppo anomalo della laringe, che provoca il pianto caratteristico del bambino. La malattia è incompatibile con la vita.

Largamente risaputo La malattia di Downè il risultato della presenza nel cariotipo di un cromosoma in più della 21a coppia (trisomia sul 21o cromosoma). Il motivo è la non-giunzione dei cromosomi sessuali durante la formazione delle cellule germinali nella madre. Nella maggior parte dei casi di comparsa di un cromosoma in più nei neonati, l'età della madre raggiunge almeno i 35 anni. Il monitoraggio della frequenza di questa malattia in aree con grave inquinamento ambientale ha rilevato un aumento significativo del numero di pazienti affetti da questa sindrome. Si presume inoltre che l'effetto di un'infezione virale sul corpo della madre durante la maturazione dell'uovo.
Una categoria separata di malattie ereditarie è sindromi associate a un cambiamento nel numero normale di cromosomi sessuali. Come la malattia di Down, si verificano quando si verifica una violazione del processo di segregazione cromosomica nella gametogenesi nella madre.
Negli esseri umani, a differenza della Drosophila e di altri animali, il cromosoma Y svolge un ruolo importante nella determinazione e nello sviluppo del sesso. In assenza di esso nell'insieme di un qualsiasi numero di cromosomi X, l'individuo sarà fenotipicamente femminile e la sua presenza determina lo sviluppo verso il sesso maschile. In particolare sono malati i maschi con il corredo cromosomico XXY+44A Sindrome di Klinefelter. Sono caratterizzate da ritardo mentale, crescita sproporzionata degli arti, testicoli molto piccoli, assenza di spermatozoi, sviluppo anomalo delle ghiandole mammarie e altri segni patologici. Un aumento del numero di cromosomi X in combinazione con un cromosoma Y non cambia la definizione di maschio, ma rafforza solo la sindrome di Klinefelter. Il cariotipo XXYY fu descritto per la prima volta nel 1962 in un ragazzo di 15 anni con significativo ritardo mentale, proporzioni corporee eunucoidi, testicoli piccoli e capelli di tipo femminile. Segni simili sono tipici dei pazienti con cariotipo XXXYY.
 Sindrome di Klinefelter (1) e sindrome di Turner-Shereshevskij (2)
Sindrome di Klinefelter (1) e sindrome di Turner-Shereshevskij (2)
L'assenza di uno dei due cromosomi X nel cariotipo femminile (XO) provoca lo sviluppo Sindrome di Turner-Shereshevskij. Le donne affette sono generalmente basse, inferiori a 140 cm, tozze, con ghiandole mammarie poco sviluppate e presentano caratteristiche pieghe pterigoidee sul collo. Di norma, sono sterili a causa del sottosviluppo del sistema riproduttivo. Molto spesso, la gravidanza con questa sindrome porta all'aborto spontaneo. Solo circa il 2% delle donne malate porta avanti la gravidanza fino alla fine.
La trisomia (XXX) o polisomia sul cromosoma X nelle donne spesso causa una malattia simile alla sindrome di Turner-Shereshevskij.
Le malattie ereditarie associate a un cambiamento nel numero dei cromosomi X vengono diagnosticate con il metodo citologico in base al numero di corpi di Barr o alla cromatina sessuale nelle cellule. Nel 1949, M. Barr e C. Bertram, studiando i nuclei interfase dei neuroni in un gatto, trovarono in essi un corpo intensamente colorato. Era presente solo nei nuclei delle cellule femminili. Si è scoperto che si verifica in molti animali ed è sempre associato al sesso. Questa struttura si chiama cromatina sessuale o corpi di Barr. Nel corso di un'accurata analisi citologica e citogenetica si è scoperto che la cromatina sessuale è uno dei due cromosomi sessuali femminili, che si trova in uno stato di forte spiralizzazione e quindi inattivo. Nelle donne con sindrome di Turner-Shereshevskij (cariotipo XO), la cromatina sessuale non viene rilevata, così come negli uomini XY normali. Le donne XX normali e gli uomini anormali hanno ciascuno un corpo Barr, mentre le donne XXX e gli uomini XXXY ne hanno due ciascuno, e così via.
Gli individui affetti da malattie ereditarie nascono solitamente con anomalie fisiche gravi, che consentono la diagnosi precoce della malattia. Ma a volte la malattia non si fa sentire per mesi e addirittura decenni. Ad esempio, una grave malattia ereditaria causata da un danno al sistema nervoso centrale - corea di Huntington- può manifestarsi solo dopo 40 anni, e quindi il suo portatore ha il tempo di lasciare la prole. I pazienti sono caratterizzati da movimenti di contrazione involontaria della testa e degli arti.
Succede che una persona dà l'impressione di un individuo assolutamente sano, ma ha una predisposizione ereditaria a una certa malattia, che si manifesta sotto l'influenza di fattori esterni o interni. Ad esempio, alcune persone hanno una reazione grave a determinati farmaci, dovuta a un difetto genetico, ovvero l'assenza di un enzima specifico nel corpo. A volte si verifica una reazione fatale all'anestesia in persone apparentemente sane, ma in realtà portano una speciale malattia muscolare ereditaria in forma latente. In tali pazienti, durante o dopo un'operazione in anestesia, la temperatura aumenta improvvisamente (fino a 42 °).
La manifestazione nell'uomo di alcune malattie ereditarie, secondo gli scienziati, è associata a diversi motivi:
- cambiamento nel numero di cromosomi;
- violazioni nella struttura dei cromosomi dei genitori;
- mutazioni a livello genetico.
Del totale, solo una coppia contiene cromosomi sessuali e tutti gli altri sono autosomici e differiscono l'uno dall'altro per dimensione e forma. Una persona sana ha 23 coppie di cromosomi. La comparsa di un cromosoma in più o la sua scomparsa provoca vari cambiamenti costituzionali nel corpo umano.
Come risultato dello sviluppo della scienza moderna, gli scienziati non solo hanno contato i cromosomi, ma ora possono riconoscere ogni coppia. L'analisi dei cariotipi consente di identificare l'esistenza di una malattia ereditaria nelle prime fasi della vita di una persona. Questi cambiamenti sono associati a uno squilibrio in una particolare coppia di cromosomi.
Cause delle malattie ereditarie
Cause delle malattie ereditarie associati a cause ereditarie possono essere suddivisi in diversi gruppi:
- malattie ad effetto diretto o congenite; compaiono nel bambino immediatamente dopo la nascita. I rappresentanti tipici includono l'emofilia, la fenilchetonuria, la malattia di Down. Gli scienziati collegano direttamente l'insorgenza di tali malattie con il modo e le condizioni di vita vissute da entrambi i genitori prima di contrarre un matrimonio congiunto e concepire un figlio. Spesso la causa dello sviluppo di questo tipo di patologia è lo stile di vita della futura mamma durante la gravidanza. Molto spesso, tra le ragioni che contribuiscono ai cambiamenti nell'insieme dei cromosomi c'è l'uso di bevande alcoliche, sostanze contenenti farmaci e condizioni ambientali negative.
- malattie ereditate dai genitori, ma attivate da una forte esposizione a stimoli esterni. Tali malattie progrediscono nel processo di crescita e sviluppo del bambino, la loro comparsa e l'ulteriore espansione provocheranno la negatività dei meccanismi responsabili dell'ereditarietà. Il principale fattore che innesca l’aumento dei sintomi è uno stile di vita socialmente negativo. Molto spesso, questi fattori possono causare diabete e disturbi mentali.
- malattie direttamente correlate alla predisposizione ereditaria. In presenza di fattori gravi associati a condizioni esterne, possono svilupparsi asma bronchiale, aterosclerosi, alcune malattie cardiache, ulcere e così via. I fattori dannosi includono un'alimentazione di scarsa qualità, un'ecologia negativa, farmaci sconsiderati, l'uso costante di prodotti chimici domestici.
Cambiamenti ereditari cromosomici

Le mutazioni associate a un cambiamento nel numero di cromosomi sembrano una violazione del processo di divisione: la meiosi. Come risultato di un fallimento nel "programma", si verifica una duplicazione delle coppie di cromosomi esistenti, sia sessuali che somatici. Le deviazioni ereditarie dipendenti dal sesso vengono trasferite utilizzando il cromosoma sessuale X.
Nel corpo maschile, questo cromosoma è senza coppia, preservando così in anticipo la manifestazione di una malattia ereditaria negli uomini. Nel corpo femminile c'è una coppia di "X", quindi le donne sono considerate portatrici del cromosoma X di bassa qualità. In modo da malattie ereditarie cromosomiche trasmesso esclusivamente per linea femminile, è necessaria la presenza di una coppia anomala. Un tale effetto è piuttosto raro in natura.
Malattie genetiche ereditarie

La maggior parte delle malattie ereditarie si verifica a causa di mutazioni genetiche, che sono cambiamenti nel DNA a livello molecolare e sono ben note agli scienziati genetici e ai pediatri. Esistono mutazioni genetiche che si manifestano a livello molecolare, cellulare, tissutale o di organo. Nonostante il fatto che l'intervallo tra una mutazione a livello delle molecole di DNA e il fenotipo principale sia ampio, va sottolineato che tutte le possibili mutazioni nei tessuti, negli organi e nelle cellule del corpo appartengono al fenotipo. Sebbene siano cambiamenti puramente esterni.
Tra le altre cose, non bisogna perdere di vista la possibilità di un impatto pericoloso dell'ecologia e di altri geni che causano varie modifiche e implementano le funzioni dei geni mutanti. Le molteplici forme delle proteine, la diversità delle loro funzioni e la mancanza di conoscenze scientifiche nel campo dei processi metabolici influiscono negativamente sui tentativi di creare una classificazione delle malattie genetiche.
Conclusione
La medicina moderna conta circa 5500-6500 forme cliniche di malattie genetiche. Questi dati sono indicativi a causa della mancanza di confini chiari nella separazione dei singoli moduli. Alcuni malattie genetiche ereditarie sono forme diverse dal punto di vista clinico, ma dal punto di vista genetico sono la conseguenza di una mutazione in un locus.
All'inizio del 21° secolo esistono già più di 6mila tipi di malattie ereditarie. Ora in molti istituti del mondo si studia una persona, il cui elenco è enorme.
La popolazione maschile ha sempre più difetti genetici e sempre meno possibilità di concepire un bambino sano. Sebbene tutte le ragioni per lo sviluppo dei difetti non siano chiare, si può presumere che nei prossimi 100-200 anni la scienza riuscirà a risolvere questi problemi.
Cosa sono le malattie genetiche? Classificazione
La genetica come scienza iniziò il suo percorso di ricerca nel 1900. Le malattie genetiche sono quelle associate ad anomalie nella struttura dei geni umani. Le deviazioni possono verificarsi sia in 1 gene che in diversi.
Malattie ereditarie:
- Autosomico dominante.
- Autosomico recessivo.
- Agganciato al pavimento.
- Malattie cromosomiche.
La probabilità di una deviazione autosomica dominante è del 50%. Con autosomico recessivo - 25%. Le malattie legate al sesso sono quelle causate da un cromosoma X danneggiato.
malattie ereditarie
Ecco alcuni esempi di malattie, secondo la classificazione di cui sopra. Quindi, le malattie dominanti-recessive includono:
- Sindrome di Marfan.
- Mioplegia parossistica.
- Talassemia.
- Otosclerosi.
Recessivo:
- Fenilchetonuria.
- Ittiosi.
- Altro.
Malattie legate al sesso:
- Emofilia.
- Distrofia muscolare.
- Malattia di Farby.
Anche sull'udito malattie ereditarie cromosomiche umane. L'elenco delle anomalie cromosomiche è il seguente:
- Sindrome di Shereshevskij-Turner.
- Sindrome di Down.
Le malattie poligeniche includono:
- Lussazione dell'anca (congenita).
- Difetti cardiaci.
- Schizofrenia.
- Labbro e palatoschisi.
L'anomalia genetica più comune è la sindattilia. Cioè, la fusione delle dita. La sindattilia è il disturbo più innocuo e viene trattato con un intervento chirurgico. Tuttavia, questa deviazione accompagna altre sindromi più gravi.
Quali malattie sono le più pericolose
Di queste malattie elencate, si possono distinguere le malattie umane ereditarie più pericolose. Il loro elenco comprende quei tipi di anomalie in cui si verifica trisomia o polisomia nel set cromosomico, cioè quando invece di una coppia di cromosomi si osserva la presenza di 3, 4, 5 o più. C'è anche 1 cromosoma invece di 2. Tutte queste deviazioni si verificano a causa di una violazione della divisione cellulare.
Le malattie ereditarie umane più pericolose:
- Sindrome di Edwards.
- Amiotrofia muscolare spinale.
- Sindrome di Patau.
- Emofilia.
- Altre malattie.
Come risultato di tali violazioni, il bambino vive per un anno o due. In alcuni casi le deviazioni non sono così gravi e il bambino può vivere fino a 7, 8 o addirittura 14 anni.
Sindrome di Down
La sindrome di Down viene ereditata se uno o entrambi i genitori sono portatori di cromosomi difettosi. Più specificamente, la sindrome è collegata a un cromosoma (vale a dire, il cromosoma 21 è 3, non 2). I bambini con sindrome di Down presentano strabismo, rughe del collo, orecchie dalla forma anomala, problemi cardiaci e ritardo mentale. Ma per la vita dei neonati un'anomalia cromosomica non rappresenta un pericolo.

Ora le statistiche dicono che su 700-800 bambini, 1 nasce con questa sindrome. Le donne che vogliono avere un bambino dopo i 35 anni hanno maggiori probabilità di avere un bambino del genere. La probabilità è di circa 1 su 375. Ma una donna che decide di avere un bambino a 45 anni ha una probabilità di 1 su 30.
acrocraniodisfalangia
Il tipo di ereditarietà dell'anomalia è autosomico dominante. La causa della sindrome è una violazione del cromosoma 10. Nella scienza, questa malattia si chiama acrocraniodisfalangia, se è più semplice, allora sindrome di Apert. È caratterizzato da caratteristiche strutturali del corpo come:
- brachicefalia (violazioni del rapporto tra larghezza e lunghezza del cranio);
- fusione delle suture coronali del cranio, a seguito della quale si osserva ipertensione (aumento della pressione sanguigna all'interno del cranio);
- sindattilia;
- fronte convessa;
- spesso ritardo mentale sullo sfondo del fatto che il cranio comprime il cervello e non consente alle cellule nervose di crescere.

Al giorno d'oggi, i bambini con la sindrome di Apert vengono sottoposti a un intervento chirurgico di aumento del cranio per ripristinare la pressione sanguigna. E il sottosviluppo mentale viene trattato con stimolanti.
Se in famiglia c'è un bambino a cui è stata diagnosticata la sindrome, la probabilità che nasca un secondo figlio con la stessa anomalia è molto alta.
La sindrome della bambola felice e la malattia di Canavan-Van Bogart-Bertrand
Diamo uno sguardo più da vicino a queste malattie. Puoi riconoscere la sindrome di Engelman da qualche parte da 3-7 anni. I bambini hanno crampi, cattiva digestione, problemi di coordinazione dei movimenti. La maggior parte di loro soffre di strabismo e problemi ai muscoli del viso, per cui molto spesso il sorriso appare sul viso. I movimenti del bambino sono molto limitati. Per i medici, questo è comprensibile quando un bambino cerca di camminare. I genitori nella maggior parte dei casi non sanno cosa sta succedendo e ancor di più con cosa è collegato. Un po 'più tardi, si nota anche che non possono parlare, cercano solo di mormorare qualcosa in modo inarticolato.

Il motivo per cui un bambino ha una sindrome è un problema nel cromosoma 15. La malattia è estremamente rara: 1 caso ogni 15mila nascite.
Un'altra malattia - la malattia di Canavan - è caratterizzata dal fatto che il bambino ha un tono muscolare debole, ha problemi con la deglutizione del cibo. La malattia è causata da un danno al sistema nervoso centrale. Il motivo è la sconfitta di un gene nel 17° cromosoma. Di conseguenza, le cellule nervose del cervello vengono distrutte con velocità progressiva.

I segni della malattia possono essere visti a 3 mesi di età. La malattia di Canavan si manifesta come segue:
- Macrocefalia.
- Le convulsioni compaiono all'età di un mese.
- Il bambino non è in grado di tenere la testa dritta.
- Dopo 3 mesi, i riflessi tendinei aumentano.
- Molti bambini diventano ciechi all’età di 2 anni.
Come puoi vedere, le malattie ereditarie umane sono molto diverse. Questo elenco è solo esemplificativo ed è lungi dall'essere completo.
Vorrei sottolineare che se entrambi i genitori hanno una violazione in 1 e nello stesso gene, allora le possibilità di dare alla luce un bambino malato sono alte, ma se ci sono anomalie in geni diversi, non c'è bisogno di aver paura. È noto che nel 60% dei casi le anomalie cromosomiche nel feto portano ad aborto spontaneo. Ma ancora il 40% di questi bambini nasce e lotta per la propria vita.
Gli scienziati sostengono che l'aspetto, lo stato di salute e altre caratteristiche individuali di una persona dipendono da due fattori principali: e l'influenza dell'ambiente. E la quota genetica rappresenta il 70%.
La maggior parte delle malattie sono in una certa misura legate all'ereditarietà: a volte, a causa della genetica, aumenta il rischio di sviluppare una determinata malattia, ma ci sono anche una serie di disturbi che sono direttamente correlati a una rottura dell'apparato genetico. Tuttavia, non tutto è perduto: ognuno di noi ha la possibilità di influenzare il proprio destino, perché il 30% della salute dipende dallo stile di vita, dalla dieta, dall'attività fisica e dagli sforzi dei medici.
Caratteristiche delle malattie trasmesse per via ereditaria
Le malattie congenite ed ereditarie non sono la stessa cosa, anche se entrambe hanno origine dal momento della nascita del bambino.
Le malattie congenite si formano a causa di una violazione del corso della gravidanza, dell'influenza di alcol, nicotina, alcuni farmaci e malattie (epatite virale). Il feto inizialmente era sano.
Le malattie con predisposizione ereditaria non lasciano al bambino nemmeno una spettrale possibilità. In questo caso, la disgregazione avviene molto prima, nella fase di trasferimento del materiale genetico dai genitori ai figli.
La seconda caratteristica dei disturbi ereditari è l'impossibilità di una cura completa. La polmonite e la tonsillite possono essere curate assumendo antibiotici, è possibile rimuovere un'appendice infiammata o una cistifellea. Ma non è ancora possibile correggere il materiale genetico. Gli scienziati stanno cercando di correggere il materiale genetico, ma è ancora lontano dall'introduzione degli sviluppi nella pratica diffusa.
L'unico modo possibile per curare le malattie ereditarie è la terapia volta ad eliminare i sintomi e migliorare la qualità della vita. In alcuni casi, la prevenzione farmacologica delle riacutizzazioni ha un effetto, ma la prognosi rimane ancora deludente. Le malattie ereditarie, purtroppo, sono ancora incurabili.
Le 5 principali malattie ereditarie
La miopia è la malattia ereditaria più comune1. Miopia
Questa è forse una delle malattie più comuni ereditate direttamente. Naturalmente anche la postura sbagliata durante la lettura, la visione frequente della TV, le molte ore trascorse quotidianamente davanti allo schermo del laptop e la mancanza di contenuti sufficienti nella dieta giocano un ruolo nel deterioramento della vista.
Tuttavia, nella stessa classe della scuola ci sono bambini che si comportano allo stesso modo, mentre uno porta già gli occhiali e l'altro vede chiaramente. La causa principale della miopia è l'ereditarietà gravata.
La causa della malattia è una caratteristica dei muscoli che contribuiscono all'estensione del bulbo oculare. Di conseguenza, l'immagine non è focalizzata sulla retina, ma più vicina e la persona vede indistintamente.
Se la madre o il padre soffrivano di miopia, la probabilità di trasmissione al bambino è del 30-40% e se entrambi sono del 70%. La malattia si manifesta spesso durante il periodo di crescita attiva - nell'adolescenza, ma anche uno studente più giovane può ammalarsi.
Questa è una classica malattia ereditaria. Esistono diverse sottospecie di emofilia, in cui la disgregazione porta a un'interruzione della produzione dei singoli fattori della coagulazione. Anche la gravità varia. Esistono tre tipi di malattia: emofilia A, B e C.
La mutazione che dà origine all’emofilia è legata al cromosoma X. Le donne hanno due cromosomi X, quindi, se uno di loro ha questa anomalia, la donna non si ammala, ma diventa semplicemente portatrice. La storia ha solo 60 casi in cui la patologia colpì due cromosomi contemporaneamente e la donna si ammalò.
Quasi tutti i pazienti affetti da emofilia sono ragazzi, perché hanno un cromosoma X. Uno degli emofiliaci più famosi fu il giovane Tsarevich Alexei Nikolaevich. Il giorno dell'esecuzione, all'età di 14 anni, il ragazzo era in condizioni estremamente gravi.
3. Trombofilia
La trombofilia è una condizione patologica in cui aumenta la coagulazione del sangue. Esistono molte varietà di trombofilia in cui si verificano mutazioni in singole parti del sistema di coagulazione (ad esempio, carenza di antitrombina, proteine C e S e sindrome da antifosfolipidi).
A molti sembra che questa condizione sia rara e non li influenzerà. E, tuttavia, è la trombofilia che spesso porta ad infarti ischemici, ictus, embolia polmonare e trombosi vascolare nelle persone sotto i 40 anni.
Spesso, la trombofilia viene rilevata durante gli esami per aborti abituali e aborti spontanei nelle donne. Sfortunatamente, è probabile che la condizione venga ereditata dai figli dei pazienti.
Questa malattia si verifica in uno su 2500 neonati, il che non è raro. La fibrosi cistica è ereditata con modalità autosomica recessiva. Cioè, affinché possa nascere un bambino malato, il bambino deve ricevere contemporaneamente il gene sbagliato dalla madre e dal padre.
Dal 2 al 5% delle persone nel mondo sono portatrici di fibrosi cistica e non ne hanno nemmeno idea. Se incontrano qualcuno come lui, possono dare alla luce un bambino malato con una probabilità del 25%.
La fibrosi cistica è associata ad una diminuzione della produzione di secrezioni da parte di tutte le ghiandole del corpo. Di conseguenza, il lavoro dei sistemi respiratorio e digestivo viene interrotto. In particolare, nessun segreto viene secreto dal lume dei bronchi in caso di malattie respiratorie, e non vi è produzione di enzimi per la digestione del cibo da parte del pancreas.
Il trattamento consiste esclusivamente nella terapia sostitutiva e la prognosi rimane sfavorevole. In Europa queste persone vivono fino a 40 anni, in Russia fino a un massimo di 28.
5. Miodistrofia
Questa terribile malattia comprende diverse sottospecie contemporaneamente (Erba-Rota, Landuzi, Duchenne). L'essenza della malattia risiede nella progressiva debolezza muscolare, che porta gradualmente alla completa immobilizzazione di una persona.
Tuttavia, dato che la malattia si trasmette con un gene recessivo, un bambino affetto da miodistrofia può nascere da genitori apparentemente sani, è sufficiente che la probabilità di avere i genitori sia del 25%.
Di norma, i medici rilevano i primi segni di miopatia di Duchenne all'età di 6 mesi. A volte vengono addirittura “cancellati” come complicanza della vaccinazione DPT, il che è fondamentalmente sbagliato, perché la malattia è ereditaria. Il modulo giovanile dell'Erba-Roth debutta a 14-16 anni.
Il trattamento della miodistrofia è sintomatico e mira esclusivamente a migliorare la qualità e massimizzare l'estensione della vita.
È possibile prevenire le malattie genetiche?
Gli scienziati non sanno ancora come curare le malattie genetiche, ma tali tentativi vengono fatti in tutto il mondo.Ad oggi è impossibile prevenire l’insorgenza di malattie ereditarie. Tuttavia, è possibile farsi esaminare per individuare i tipi più comuni di mutazioni e identificare la probabilità di avere un figlio affetto da una patologia in una determinata coppia.
Molto dipende dal comportamento dei genitori. Il cambiamento dello stile di vita, ovviamente, non influirà sul materiale genetico, ma in alcuni casi riduce il rischio di gravi manifestazioni della malattia.
Pertanto, non abbiate paura dei test genetici: prima viene fatta la diagnosi, più facile sarà aiutare il bambino. Se non sai a quale laboratorio rivolgerti ti selezioneranno gratuitamente un laboratorio dove potrai effettuare un test genetico per le malattie congenite ad un prezzo conveniente.
9.1 Il concetto, la classificazione e le caratteristiche della patologia ereditaria
La patologia è qualsiasi deviazione dal normale corso dei processi biologici: metabolismo, crescita, sviluppo, riproduzione.
La patologia ereditaria è una deviazione dalla norma con un fatto accertato di eredità, cioè trasmissione di generazione in generazione. È necessario distinguere tra una patologia congenita – presente fin dalla nascita di un individuo – da una patologia ereditaria. La patologia congenita può essere causata dall'azione di fattori ambientali: mancanza di nutrienti e ossigeno durante lo sviluppo fetale, lesioni alla nascita, infezioni e così via. L'accertamento dell'ereditarietà di un tratto anormale secondo i requisiti dell'analisi genetica (capitolo II) è l'unica base per riconoscere la natura ereditaria della patologia.
Esistono due tipi di classificazione della patologia ereditaria. Il primo (accettato principalmente nella letteratura nazionale) è di tipo clinico. Secondo questo tipo di classificazione si distinguono quattro gruppi di malattie:
Gruppo I - si tratta in realtà di malattie ereditarie - malattie cromosomiche e genetiche (sindromi di Edwards e Patau, fenilchetonuria, fibrosi cistica);
Gruppo II - malattie con una predisposizione ereditaria pronunciata, nella cui patogenesi la manifestazione di fattori ereditari è determinata dall'azione di specifiche circostanze esterne (ipertensione arteriosa, diabete mellito, gotta);
Gruppo III - malattie determinate principalmente da fattori ambientali, ma nella patogenesi delle quali i fattori ereditari svolgono un certo ruolo (glaucoma, aterosclerosi, cancro al seno);
Gruppo IV - malattie a cui l'ereditarietà a prima vista non è correlata (intossicazione alimentare, fratture, ustioni).
Va notato che i concetti frequentemente utilizzati di malattie "familiari" e "sporadiche" non sono direttamente correlati all'ereditarietà. Le malattie familiari si osservano nei parenti, ma possono anche essere causate dall'azione delle stesse cause esterne, ad esempio la natura della nutrizione. Casi sporadici si verificano in singoli individui, ma possono anche essere dovuti a una rara combinazione di alleli o a una mutazione de novo.
Il secondo sistema di classificazione - genetico - è generalmente accettato nella letteratura straniera e recentemente è stato sempre più utilizzato nella letteratura russa. Secondo questo sistema si distinguono cinque gruppi:
Gruppo I: malattie genetiche determinate da mutazioni in alcuni geni. Si tratta di tratti prevalentemente monogenici con modelli di ereditarietà autosomica dominante, autosomica recessiva, dominante legata al sesso, recessiva legata al sesso, olandese e mitocondriale (Capitolo II);
Gruppo II - malattie cromosomiche, cioè mutazioni genomiche e cromosomiche (Capitolo V);
Gruppo III - malattie con predisposizione ereditaria, nella patogenesi delle quali giocano un ruolo fattori ambientali ed ereditari, aventi un tipo di eredità monogenica o poligenica (miopia, obesità patologica, ulcere allo stomaco).
Gruppo IV - malattie genetiche delle cellule somatiche, spesso associate a neoplasie maligne (retinoblastoma, tumore di Wilms, alcune forme di leucemia);
Gruppo V - malattie di incompatibilità genetica della madre e del feto, che si sviluppano a seguito della risposta immunitaria della madre agli antigeni fetali (incompatibilità per il fattore Rh e alcuni altri sistemi antigene-anticorpo eritrocitario).
Le malattie ereditarie possono iniziare a manifestarsi in età diverse. La natura della manifestazione (il momento della manifestazione dei primi sintomi della malattia) è specifica per le diverse forme di patologia ereditaria. Di norma, le malattie ereditarie sono caratterizzate da un decorso cronico (a lungo termine) progressivo (con un aumento della gravità dei sintomi).
9.2 Malattie cromosomiche
Questo gruppo comprende malattie causate da anomalie nel numero o nella struttura dei cromosomi. Circa l'1% dei neonati presenta un cariotipo anomalo e, tra i nati morti, l'incidenza di aberrazioni nel numero o nella struttura dei cromosomi è del 20%. I tratti caratteristici comuni delle malattie cromosomiche sono: basso peso alla nascita, ritardo dello sviluppo, bassa statura, microcefalia, micrognazia, disturbi dell'osteogenesi, posizione anormale degli occhi. Una descrizione più dettagliata delle malattie cromosomiche è fornita nelle sezioni 5.8 e 5.9.
9.3 Malattie genetiche
Le malattie genetiche sono condizioni patologiche causate da mutazioni genetiche. Molto spesso, questo concetto viene applicato alle malattie monogeniche.
Questo gruppo è caratterizzato da eterogeneità: le stesse malattie possono essere causate da mutazioni in geni diversi. I principi generali per lo sviluppo della patologia a livello genetico possono essere:
Produzione di un prodotto proteico anomalo;
Assenza di proteine normali;
Quantità insufficiente di proteine normali;
Un eccesso di un normale prodotto proteico.
In base alla natura delle violazioni dell'omeostasi (la costanza dell'ambiente interno del corpo), si distinguono i seguenti gruppi di malattie genetiche:
1. Malattie del metabolismo degli aminoacidi.
Il gruppo più numeroso di malattie metaboliche ereditarie. Quasi tutti vengono ereditati con modalità autosomica recessiva. La causa delle malattie è l'insufficienza dell'uno o dell'altro enzima responsabile della sintesi degli aminoacidi.
Fenilchetonuria- violazione della conversione della fenilalanina in tirosina a causa di una forte diminuzione dell'attività della fenilalanina idrossilasi - una malattia autosomica recessiva. Appare all'età di 2-4 mesi, i primi sintomi sono letargia, convulsioni, eczema, odore di "topo" (l'odore dei chetoni). Si sviluppa gradualmente un grave danno cerebrale, che porta a una forte diminuzione dell'intelligenza fino all'idiozia. Se fin dai primi giorni di vita si esclude completamente (o si limita significativamente la quantità) la fenilalanina dalla dieta di un bambino malato prima della pubertà, i sintomi non si sviluppano. La malattia è causata da mutazioni nel gene PAH, che codifica per la fenilalanina-4-idrossilasi. Gene PAH localizzato in HSA12q24.1. Diverse dozzine di mutazioni di questo gene sono state descritte in diverse popolazioni. Esistono sistemi diagnostici basati sulla PCR in grado di rilevare il portatore eterozigote. Recentemente sono stati sviluppati nuovi approcci al trattamento della fenicetonuria: terapia sostitutiva con fenilalanina liasi, un enzima vegetale che catalizza la scomposizione della fenilalanina in metaboliti innocui, e terapia genica inserendo un normale gene della fenilalanina idrossilasi nel genoma.
Alcaptonuria- disturbo autosomico recessivo del metabolismo della tirosina e accumulo nei tessuti corporei (cartilagine articolare, tendini) di acido omogentisico. La manifestazione avviene durante l'infanzia. Il primo sintomo è l'urina scura. Spesso si sviluppa urolitiasi e pielonefrite. L'accumulo dei prodotti di degradazione dell'acido omogentisico porta a danni alle articolazioni (principalmente ginocchio e anca). C'è un oscuramento e una maggiore fragilità del tessuto connettivo. È caratteristico l'oscuramento della sclera e dei padiglioni auricolari. Mutazioni in un gene HGD- le ossidasi dell'acido omogentisico sono la causa di questa malattia. Questo gene contiene 14 esoni e si trova in HSA3q21-23. Sono state descritte circa 100 diverse mutazioni missenso, mutazioni di tipo frameshift e cambiamenti nel sito di giunzione associati a questa malattia. .
Albinismo oculocutaneo 1- assenza o significativa mancanza di pigmento nella pelle, nei capelli, nell'iride e nelle membrane pigmentate dell'occhio (Figura IX, 1).
Figura IX, 1. Il rappresentante della razza negroide è un albino. Basato su materiali dal sito http://upload.wikimedia.org/wikipediacommons/99a/Albinisitic_man_ Portrait
Una malattia con modalità di trasmissione autosomica recessiva. Si manifesta con vari gradi di depigmentazione della pelle, dei capelli, dell'iride e delle membrane pigmentate dell'occhio, diminuzione dell'acuità visiva, fotofobia, nistagmo e frequenti scottature solari. Varie mutazioni missenso, mutazioni di tipo frameshift e mutazioni non senso nel gene della tirosinasi ( TIR, HSA11q24) sono responsabili di questa malattia.
2. Disturbi del metabolismo dei carboidrati
Galattosemia- l'assenza o una significativa diminuzione dell'attività dell'enzima galattosio-1-fosfato-uridiltransferasi e l'accumulo di galattosio e dei suoi derivati nel sangue, che hanno un effetto tossico sul sistema nervoso centrale, sul fegato e sul cristallino dell'occhio. Nei primi giorni e settimane di vita si osservano ittero, ingrossamento del fegato, nistagmo, ipotonia muscolare e vomito. Nel corso del tempo si sviluppa una cataratta, un ritardo nello sviluppo fisico e mentale. Caratterizzato da intolleranza al latte.
La malattia ha una modalità di trasmissione autosomica recessiva. Diverse forme di questa malattia sono causate da diversi alleli mutanti del gene GAL(galattosio-1-fosfato uridiltransferasi), localizzato nella regione di HSA9p13. Le mutazioni missenso riducono l'attività dell'enzima a vari livelli, il che determina la diversa gravità dei sintomi della malattia. Ad esempio, la galattosemia di Durte è quasi asintomatica, si nota solo una tendenza ai disturbi epatici.
Malattia di Gierke (glicogenosi di tipo I, glicogenosi di tipo I)- l'incapacità di convertire il glucosio-6-fosfato in glucosio, che porta a una violazione della sintesi e della decomposizione del glicogeno. Si verifica la deposizione di glicogeno, il processo inverso no. Si sviluppa ipoglicemia. L'accumulo di glicogeno in eccesso nel fegato e nei reni porta a insufficienza epatica e renale. Il tipo di ereditarietà è autosomica recessiva. La causa della malattia è una mutazione nel gene G6PC, che codifica per l'enzima glucosio-6-fosfatasi. Sono stati descritti 14 alleli mutanti di questo gene e sono associati alla malattia di Gierke. Esistono test genetici molecolari per individuare il portatore eterozigote e la diagnosi prenatale di questa malattia.
3. Disturbi del metabolismo dei lipidi
Malattia di Niemann-Pick di tipo A e B- diminuzione dell'attività dell'enzima sfingomielinasi lisosomiale acida, codificato dal gene SMPD1(HSA11p15.4-p15.1). Il tipo di ereditarietà è autosomica recessiva. La violazione del metabolismo dei lipidi porta all'accumulo di lipidi nel fegato, nei polmoni, nella milza e nei tessuti nervosi. Caratterizzato da degenerazione delle cellule nervose, interruzione del sistema nervoso, elevati livelli di colesterolo e lipidi nel sangue. Il tipo A è letale nella prima infanzia. Il tipo B è più lieve e i pazienti di solito sopravvivono fino all’età adulta. Diversi tipi sono causati da diverse mutazioni nel gene SMPD1.
Malattia di Gaucher (lipidosi da glicosilceramide)- accumulo di glucocerebrosidi nelle cellule del sistema nervoso e reticoloendoteliale, a causa di una carenza dell'enzima glucocerebrosidasi, codificato dal gene GBA(HSA1q21). Appartiene al gruppo delle malattie da accumulo lisosomiale. Alcune forme della malattia si manifestano con gravi lesioni del fegato, della milza, dei tessuti nervosi e ossei.
4. Malattie ereditarie del metabolismo delle purine e delle pirimidine
Sindrome di Lesch-Nychen - una malattia recessiva legata al sesso in cui il contenuto di acido urico in tutti i fluidi corporei aumenta notevolmente. La conseguenza di ciò è un ritardo dello sviluppo, un moderato ritardo mentale, attacchi di comportamento aggressivo con autolesionismo. Insufficienza dell'attività enzimatica dell'ipoxantina-guanina fosforibosiltransferasi dovuta a mutazioni nel gene HPRT1(HSAXq26-q27.2) è alla base di questa malattia. Sono state descritte diverse mutazioni nello stesso gene, che hanno portato a gotta(violazione del metabolismo delle purine e deposizione di composti di acido urico nei tessuti).
5. Disturbi del metabolismo del tessuto connettivo
Sindrome di Marfan ("dita di ragno", aracnodattilia)- danno al tessuto connettivo dovuto a una mutazione nel gene FBN1(HSA15q21.1), responsabile della sintesi della fibrillina. Si trasmette con modalità autosomica dominante. Il polimorfismo clinico della malattia è spiegato da un gran numero di alleli mutanti, ognuno dei quali può manifestarsi in uno stato eterozigote. I pazienti sono caratterizzati da crescita elevata, costituzione astenica (arti sproporzionatamente lunghi), aracnodattilia (dita lunghe e sottili), debolezza dell'apparato legamentoso, distacco della retina, sublussazione del cristallino, prolasso della valvola mitrale (Figura IX, 2).

Figura IX, 2. Sindrome di Marfan. Basato su materiali dal sito http://www.spineinfo.ru/infosources/case/cases_14.html.
Mucopolisaccaridosi- un gruppo di malattie del tessuto connettivo associate ad un alterato metabolismo dei glicosaminoglicani acidi (mucopolisaccaridi) causato da una carenza di alcuni enzimi lisosomiali. Queste malattie sono chiamate malattie da accumulo lisosomiale. Si manifestano in vari difetti delle ossa e dei tessuti connettivi. Mucopolisazidosi di tipo I (sindrome di Hurler)- una malattia autosomica recessiva derivante da un deficit dell'enzima alfa-L-iduronidasi dovuto a mutazioni nel gene IDUA (HSA4q16.3). Ciò porta all'accumulo di complessi proteine-carboidrati e grassi nelle cellule del corpo. Di conseguenza, i pazienti presentano bassa statura, ritardo mentale significativo, ingrossamento del fegato e della milza, difetti cardiaci, annebbiamento della cornea, deformità ossea e ingrossamento dei tratti facciali (Figura IX, 3).

Figura IX, 3. Sindrome di Hurler. Adattato da http://medgen.genetics.utah.edu/photographs/pages/hurler_syndrome.htm.
Mucopolisaccaridosi di tipo II(Sindrome di Hunter) è una malattia recessiva legata al sesso causata da un difetto dell'enzima iduronato solfatasi dovuto a una mutazione nel gene IDS (HSAXq28). Le sostanze di accumulo sono dermatan ed eparan solfati. Caratterizzato da tratti facciali grossolani, scafocefalia, respiro rumoroso, voce profonda e ruvida, frequenti infezioni virali respiratorie acute (Figura IX, 4 ) . All'età di 3-4 anni si verificano disturbi della coordinazione dei movimenti: l'andatura diventa goffa, i bambini spesso cadono quando camminano. I pazienti sono caratterizzati da labilità emotiva e aggressività. Si osservano anche perdita progressiva dell'udito, lesioni cutanee nodulari della schiena, artrosi, lesioni corneali.
\ 
Figura IX, 4. Sindrome del cacciatore. Basato su materiali dal sito http://1nsk.ru/news/russia/23335.html.
Mucopolisaccaridosi di tipo III (sindrome di Sanfilippo, malattia di Sanfilippo) - malattia causata dall'accumulo di eparan solfato. È caratterizzata da eterogeneità genetica: esistono 4 tipi di questa malattia causata da mutazioni in 4 diversi geni che codificano per enzimi coinvolti nel metabolismo della sostanza accumulata. I primi sintomi della malattia sotto forma di disturbi del sonno compaiono nei bambini di età superiore ai 3 anni. L'apatia si sviluppa gradualmente, c'è un ritardo nello sviluppo psicomotorio, i disturbi del linguaggio, i tratti del viso diventano ruvidi. Col passare del tempo, i bambini smettono di riconoscere gli altri. Per i pazienti sono tipici il ritardo della crescita, le contratture articolari, l'ipertricosi e l'epatosplenomegalia moderata. A differenza delle sindromi di Hurler e Hunter, nella malattia di Sanfilippo predomina il ritardo mentale e non sono presenti lesioni della cornea e del sistema cardiovascolare.

Figura IX, 5. Sindrome di Sanfilippo. Adattato da http://runkle-science.wikispaces.com/Sanfilippo-syndrome.
Fibrodisplasia (miosite ossificante, ossificazione eterotopica paraossale, malattia di Münheimer)- una malattia del tessuto connettivo associata alla sua progressiva ossificazione a seguito di una mutazione nel gene ACVR1(HSA2q23-q24), che codifica per il recettore dell'attivina A. La modalità di trasmissione è autosomica dominante. La malattia si manifesta con difetti congeniti dello sviluppo - principalmente alluci ricurvi e disturbi nella colonna cervicale a livello delle vertebre C2 - C7. La malattia ha un carattere progressivo, porta a disturbi significativi dello stato funzionale del sistema muscolo-scheletrico, grave disabilità dei pazienti e morte, principalmente nell'infanzia e nella giovane età (Figura IX, 6). La malattia è anche chiamata la “malattia del secondo scheletro”, poiché dove dovrebbero verificarsi regolari processi antinfiammatori nel corpo, inizia la crescita ossea.

Figura IX, 6. Fibrodisplasia. Basato su materiali dal sito http://donbass.ua/news/health/2010/02/15.
6. Disturbi delle proteine circolanti
Emoglobinopatie- disturbi ereditari della sintesi dell'emoglobina. Esistono due gruppi di emoglobinopatie. Il primo è caratterizzato da un cambiamento nella struttura primaria della proteina globina, che può essere accompagnato da violazioni della sua stabilità e funzione (ad esempio, anemia falciforme). Con le emoglobinopatie del secondo gruppo, la struttura dell'emoglobina rimane normale, solo la velocità di sintesi delle catene globiniche è ridotta (ad esempio, β -talassemia).
7. Disturbi metabolici negli eritrociti
sferocitosi ereditaria- deficit congenito dei lipidi dell'involucro eritrocitario. La malattia è caratterizzata da una modalità di trasmissione autosomica dominante o autosomica recessiva, a seconda della mutazione genetica SPTA1(HSA1q21), che codifica la spettrina α-1 degli eritrociti. Un'anomalia di questa proteina porta ad un aumento della concentrazione di ioni sodio all'interno dell'eritrocito e alla penetrazione di acqua in eccesso al suo interno a causa di un aumento della pressione osmotica. Di conseguenza, si formano eritrociti sferici: sferociti che, a differenza degli eritrociti normali biconcavi, non hanno la capacità di cambiare forma in sezioni strette del flusso sanguigno, ad esempio quando passano nei seni della milza. Ciò porta ad un rallentamento dell'avanzamento degli eritrociti nei seni della milza e alla scissione di una parte della membrana eritrocitaria con formazione di microsferociti. Gli eritrociti distrutti vengono assorbiti dai macrofagi nella milza. L'emolisi degli eritrociti porta all'iperplasia delle cellule della polpa e all'ingrossamento della milza. Uno dei principali sintomi clinici è l'ittero. I principali sintomi della sferocitosi ereditaria sono l'ingrossamento della milza (di solito sporgente da sotto l'ipocondrio di 2-3 cm) e l'ittero. A volte ci sono segni di sviluppo lento, violazioni dello scheletro facciale, un cranio imponente, un naso a sella, un palato alto, una violazione della posizione dei denti, orbite strette.
8. Malattie ereditarie del metabolismo dei metalli
Malattia di Konovalov-Wilson (distrofia epatocerebrale)- una malattia autosomica recessiva del metabolismo del rame, che porta a gravi lesioni del sistema nervoso centrale e degli organi interni. La malattia è causata dalla sintesi bassa o anormale di ceruloplasmina (proteina che trasporta il rame) a causa dell'attività enzimatica insufficiente dell'ATPasi che trasporta il rame. Mutazioni (ne sono state descritte circa 200) nel gene ATP7B(HSA13q14-q21) portano a cambiamenti nel β-polipeptide di questo enzima, che è la base genetica di questa patologia. Il ruolo principale nella patogenesi è giocato da una violazione del metabolismo del rame, dal suo accumulo nei tessuti nervoso, renale, epatico e nella cornea, con conseguente danno tossico a questi organi da parte del rame. Nel fegato si forma una cirrosi nodulare grande o mista. Nei reni, i tubuli prossimali sono i primi ad essere colpiti. Nel cervello sono colpiti in misura maggiore i gangli della base, il nucleo dentato del cervelletto e la substantia nigra.
9. Malassorbimento nel tratto digestivo
Fibrosi cistica (fibrosi cistica) - una malattia autosomica recessiva caratterizzata da danni alle ghiandole secretrici esterne, gravi disturbi delle funzioni dell'apparato respiratorio e del tratto gastrointestinale. Causato da mutazioni nel gene CFTR(HSA7q31.2), che codifica per un regolatore transmembrana della fibrosi cistica. La malattia è caratterizzata da danni alle ghiandole secrezionali esterne, gravi disturbi delle funzioni dell'apparato respiratorio e del tratto gastrointestinale.
Intolleranza al lattosio (ipolactasia) - una condizione patologica autosomica recessiva di cattiva digestione del lattosio (zucchero del latte), la cui base genetica sono mutazioni nelle regioni regolatrici e codificanti del gene LCT(HSA2q21), che codifica per la lattasi. Questo enzima è espresso prevalentemente nelle cellule ciliari intestinali ed è responsabile della scomposizione del lattosio in galattosio e glucosio. I principali sintomi della carenza di lattasi sono flatulenza, dolore addominale, diarrea e vomito. Nei bambini, la carenza di lattasi può manifestarsi con stitichezza cronica, irrequietezza e pianto dopo i pasti. In diverse popolazioni umane, le frequenze degli alleli mutanti variano dall'1 al 100%.
10. Disturbi ormonali
Femminilizzazione testicolare (sindrome di Morris) - una malattia recessiva legata al sesso in cui un cariotipo femminile (46,XY) mostra un fenotipo femminile. l'espressività varia. Con la femminilizzazione incompleta, le gonadi si sviluppano secondo uno schema maschile, ma alcuni caratteri sessuali corrispondono al sesso femminile con vari gradi di gravità: clitoride ipertrofico, chiusura incompleta della sutura scrotale, grandi labbra a forma di scroto, una vagina accorciata (Figura IX , 7). Con la femminilizzazione completa, il sintomo principale è l'assenza di mestruazioni e crescita dei peli sessuali con ghiandole mammarie ben sviluppate e un fenotipo femminile. La malattia è causata da varie mutazioni nel gene AR(HSAXq11-q12), che codifica per il recettore degli androgeni.

Figura IX, 7. Veduta dei genitali esterni con femminilizzazione testicolare incompleta. Basato su materiali dal sito http://www.health-ua.org/img/woman/tabl/8_17.jpg.
Sindrome androgenitale (pseudoermafroditismo femminile) - una malattia endocrina con un tipo di ereditarietà autosomica recessiva, in cui il paziente ha genitali esterni di tipo maschile e una struttura ormonale femminile. Nei pazienti, il clitoride è ingrandito, che diventa simile a un pene maschile con un'apertura urogenitale, non c'è ingresso esterno alla vagina, le piccole labbra sono assenti, le labbra grandi sembrano uno scroto "tritato". In questo caso, gli organi genitali interni possono avere un aspetto normale. La base genetica della malattia sono le mutazioni genetiche CYP21(HSA6q21.3), che codifica per l'enzima 21-idrossilasi del gruppo del citocromo P450, coinvolto nella sintesi degli ormoni aldosterone e cortisolo.
9.4 Marcatori molecolari nello studio della patologia ereditaria
Una parte significativa delle malattie ereditarie e delle malattie con predisposizione ereditaria non sono di natura monogenica. Possono essere attribuiti a tratti quantitativi, cioè quelli che presentano una serie continua di variabilità e possono essere misurati, ad esempio altezza, peso, lunghezza degli arti. Gli alleli di un gran numero di geni contribuiscono alla manifestazione di tali tratti, quindi sono chiamati poligenici. È possibile tracciare la loro eredità e identificare i geni i cui alleli sono coinvolti nei processi patologici utilizzando marcatori genetici. L'individuazione dell'eredità concatenata (associazione) di tratti fenotipici con marcatori genetici consente di individuare regioni dei cromosomi che hanno un'influenza decisiva sui processi studiati (clonazione posizionale) e di ottenere sistemi affidabili per la diagnostica molecolare (etichettatura molecolare). Attualmente, i marcatori più comuni nella genetica umana sono i loci microsatelliti (Figura IX, 8; sezione 8.1) e i siti polimorfici mononucleotidici - SNP (Figura IX, 9), le cui caratteristiche principali sono mostrate nella Tabella IX, 1.
L'analisi dell'espressione genica (di tutti o di un gruppo) su biochip nei tessuti correlati ad una particolare malattia ereditaria, in condizioni normali e patologiche, consente spesso di identificare geni candidati per la malattia in studio. La localizzazione cromosomica delle sequenze di DNA che influenzano un tratto quantitativo (QTL) può essere determinata in base alla coereditarietà con diversi marcatori ravvicinati. Se è possibile trovare marcatori che limitano il QTL su entrambi i lati, allora, sulla base dei dati di sequenziamento genomico (Sezioni 7.7 e 8.4), è possibile compilare un elenco di geni che sono candidati posizionali per il QTL della malattia sotto studio. Con l'uso simultaneo dell'analisi dell'espressione e dello studio delle associazioni di malattie con marcatori molecolari, è possibile determinare i geni candidati più probabili, quelli che compaiono in entrambi gli elenchi.
Il grado di suscettibilità a determinati farmaci e l’efficacia del loro utilizzo varia ampiamente. Per la stessa malattia, il farmaco appropriato per un particolare individuo viene spesso selezionato per tentativi ed errori. Oltre a far perdere tempo, questo approccio a volte provoca danni irreparabili alla salute. Attualmente, per un gran numero di farmaci, sono stati sviluppati sistemi di marcatori basati su SNP che consentono di prevedere a priori (prima dell'esperienza) la risposta di un singolo organismo ad una particolare sostanza chimica. Le associazioni delle singole varianti alleliche dei marcatori del DNA con le caratteristiche delle reazioni biochimiche sono la base della terapia individuale (Figura IX, 10).

Figura IX, 8. Nei loci microsatelliti, l'unità di variabilità è un gruppo di nucleotidi.

Figura IX, 9. Nei siti polimorfici mononucleotidici (SNP), l'unità di variazione è un nucleotide.
Tabella IX, 1. Confronto delle principali caratteristiche di SNP e microsatelliti.

Figura IX, 10. Il principio di selezione della terapia individuale basato sul polimorfismo ripetuto del mononucleotide - SNP.
Controlla le domande e le attività per il capitolo IX
1. Quale gruppo di malattie ereditarie può essere attribuito alla fibrosi cistica?
2. Può un eterozigote per una mutazione genetica SPTA1 essere sferocitosi ereditaria?
3. Quale malattia ereditaria è causata dall'accumulo di eparan solfato?
4. Perché esistono quattro possibili alleli SNP?
Letture aggiuntive per il capitolo IX
N.P. Bochkov. Genetica clinica // M.: Geotar-Med. 2002. - 457 pag.
- In contatto con 0
- Google+ 0
- OK 0
- Facebook 0






