
Una malattia causata da un disturbo del metabolismo proteico, in cui la formazione e la deposizione di una specifica sostanza proteica-polisaccaridica - l'amiloide - avviene in vari tessuti e organi.
Sviluppo della malattia
L'amiloidosi si sviluppa (abbiamo già scoperto di cosa si tratta) quando la sintesi proteica nel sistema reticoloendoteliale viene interrotta. Si verifica un accumulo di proteine anomale. Queste proteine sono essenzialmente autoantigeni e, analogamente alle allergie, provocano la formazione di autoanticorpi.
Quindi questi anticorpi reagiscono con gli antigeni e le proteine grossolanamente disperse precipitano. Ecco come si forma l'amiloide. Questa sostanza si deposita sulle pareti vascolari e su vari organi. Accumulandosi gradualmente, l'amiloide porta alla morte degli organi.
Tipi di amiloidosi. Cause
Esistono diversi tipi di amiloidosi. Le cause dello sviluppo della malattia dipendono direttamente dal tipo di amiloidosi. Cos'è? La classificazione si basa sulla principale proteina di cui sono composte le fibrille amiloidi. Di seguito sono riportati i tipi di questa malattia.
- Amiloidosi primaria (amiloidosi AL). Con il suo sviluppo, nel plasma sanguigno compaiono catene leggere anormali di immunoglobuline, che possono depositarsi in una varietà di tessuti del corpo. Le plasmacellule cambiano allo stesso modo nella macroglobulinemia di Waldenström e nell'ipergammaglobulinemia monoclonale.
- Amiloidosi secondaria (amiloidosi AA). In questo caso, il fegato produce una quantità eccessiva di proteina alfa-globulina. Si tratta di una proteina della fase acuta che viene sintetizzata durante un processo infiammatorio cronico. Ciò è possibile per varie malattie, ad esempio l'artrite reumatoide, la malaria, l'osteomielite, la lebbra, la tubercolosi.
- Amiloidosi familiare (amiloidosi AF). Si tratta di una forma ereditaria della malattia con un meccanismo di trasmissione autosomica recessiva. Viene anche chiamata febbre intermittente mediterranea o polisierosite parossistica familiare. Questa malattia è espressa da attacchi di febbre, dolore addominale, eruzioni cutanee, artrite e pleurite.
- Amiloidosi da dialisi (amiloidosi AH). Ciò è dovuto al fatto che la proteina beta-2-microglobulina MHC nelle persone sane viene utilizzata dai reni, ma durante l'emodialisi non viene filtrata e quindi si accumula nel corpo.
- Amiloidosi EA. Si sviluppa in alcune forme di cancro, come il cancro alla tiroide.
- Amiloidosi senile.

Sintomi
Quando viene diagnosticata l'amiloidosi, i sintomi dipendono dalla posizione dei depositi. Se è interessato il tratto gastrointestinale, possono verificarsi lingua ingrossata, compromissione della funzione di deglutizione, stitichezza o diarrea. A volte sono possibili depositi di amiloide simili a tumori nell'intestino o nello stomaco.
L'amiloidosi intestinale è accompagnata da una sensazione di pesantezza e disagio e può verificarsi un dolore moderato nella zona addominale. Se è colpito il pancreas, sono presenti gli stessi sintomi della pancreatite. Quando il fegato è danneggiato, si osserva il suo ingrossamento, compaiono nausea, eruttazione, vomito e ittero.
L'amiloidosi respiratoria si manifesta come segue:
- voce rauca;
- sintomi di bronchite;
- amiloidosi polmonare simile a un tumore.
Con l'amiloidosi del sistema nervoso, si possono osservare i seguenti sintomi:
- sensazione di formicolio o bruciore agli arti, sensazione di intorpidimento (polineuropatia periferica);
- mal di testa, vertigini;
- disturbi dello sfintere (incontinenza urinaria e fecale).
Amiloidosi: cos'è, le cause della sua comparsa e i sintomi che abbiamo esaminato. Ora scopriamo come viene diagnosticata questa malattia e quali metodi di trattamento esistono.

Diagnostica
Per una malattia come l’amiloidosi, la diagnosi è complessa. Sono prescritti esami di laboratorio e strumentali.
Negli esami di laboratorio, un esame del sangue generale mostra un aumento della VES, dei leucociti e una diminuzione delle piastrine. Nell'analisi generale delle urine sono presenti proteine, nel sedimento sono presenti cilindri, leucociti ed eritrociti. Il coprogramma contiene una grande quantità di amido, grasso e fibre muscolari. La biochimica del sangue con danno epatico rivela un aumento dei livelli di colesterolo, bilirubina e fosfatasi alcalina.
Nell'amiloidosi primaria, vengono rilevati alti livelli di amiloide nelle urine e nel plasma sanguigno. Nei casi secondari, gli esami di laboratorio rivelano segni di un processo infiammatorio cronico.
Vengono eseguite anche altre misure diagnostiche:
- esame radiografico;
- ecocardiografia (in caso di sospetto danno cardiaco);
- test funzionali con coloranti;
- biopsia d'organo.

Trattamento
Questa malattia viene trattata in regime ambulatoriale. L'amiloidosi, in cui si osservano condizioni gravi, ad esempio con insufficienza renale cronica o grave insufficienza cardiaca, viene curata in ospedale.
In caso di amiloidosi primaria, nella fase iniziale, vengono prescritti farmaci come la clorochina, il melfalan, il prednisolone e la colchicina.
Con l'amiloidosi secondaria, viene trattata la malattia di base, ad esempio osteomielite, tubercolosi, empiema pleurico, ecc. Spesso, dopo la guarigione, tutti i sintomi dell'amiloidosi scompaiono.
Se la malattia si sviluppa a seguito dell'emodialisi renale, tale paziente viene trasferito alla dialisi peritoneale.
In caso di diarrea vengono utilizzati farmaci astringenti, ad esempio subnitrato di bismuto o agenti adsorbenti.
Viene anche utilizzato il trattamento sintomatico:
- farmaci che abbassano la pressione sanguigna;
- vitamine, diuretici;
- trasfusione di plasma, ecc.
Inoltre, è possibile utilizzare il trattamento chirurgico. L'amiloidosi della milza può regredire dopo la rimozione dell'organo. Nella maggior parte dei casi, ciò porta ad un miglioramento delle condizioni dei pazienti e ad una diminuzione della formazione di amiloide.
Nutrizione
L’amiloidosi richiede una dieta costante. Se si sviluppa un'insufficienza renale cronica, è necessario limitare l'assunzione di sale e cibi proteici come carne, pesce e uova. Se si sviluppa una malattia cronica, i cibi salati, affumicati e in salamoia dovrebbero essere esclusi dalla dieta.

Questa malattia è anche chiamata cardiopatia amiloide. Durante il suo sviluppo, la deposizione di amiloide può verificarsi nel miocardio, nel pericardio, nell'endocardio o sulle pareti dell'aorta e dei vasi coronarici. La causa di tale danno cardiaco può essere l'amiloidosi primaria, secondaria o familiare. Spesso l'amiloidosi cardiaca non è una malattia isolata e si sviluppa parallelamente all'amiloidosi dei polmoni, dei reni, dell'intestino o della milza.
Sintomi dell'amiloidosi cardiaca
Spesso i sintomi di questa malattia sono simili alla cardiopatia ipertrofica o alla malattia coronarica. Nella fase iniziale, i sintomi non sono chiaramente espressi. Possono verificarsi irritabilità e affaticamento, perdita di peso, gonfiore dei tessuti e vertigini.

Un netto peggioramento di solito si verifica dopo situazioni stressanti o un'infezione respiratoria. Successivamente compaiono solitamente dolori al cuore come angina pectoris, aritmie, edema grave, mancanza di respiro e ingrossamento del fegato. La pressione sanguigna è solitamente bassa.
La malattia progredisce rapidamente e la sua caratteristica distintiva è la resistenza alla terapia. Nei casi più gravi, i pazienti possono manifestare ascite (accumulo di liquido o versamento pericardico). A causa degli infiltrati di amiloide, si sviluppano debolezza del nodo del seno e bradicardia. Ciò può portare alla morte improvvisa.
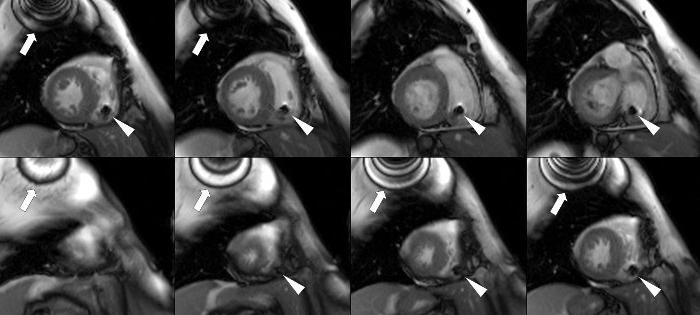
Previsione
L’amiloidosi cardiaca ha una prognosi sfavorevole. L'insufficienza cardiaca in questa malattia progredisce costantemente e la morte è inevitabile. In Russia non esistono centri specializzati che si occupano di questo problema.
Amiloidosi- un gruppo di malattie caratterizzate da un'ampia varietà di manifestazioni cliniche e caratterizzate dalla deposizione extracellulare di proteine fibrillari patologiche insolubili negli organi e nei tessuti.Amiloideè una glicoproteina complessa in cui le proteine fibrillare e globulari sono strettamente associate ai polisaccaridi. Utilizzando la microscopia elettronica e i metodi di diffrazione dei raggi X, viene chiaramente mostrata la struttura ordinata dell'amiloide e la presenza di strutture fibrose in essa. Le dimensioni delle fibrille hanno un diametro compreso tra 7,5 e 10 nm e una lunghezza fino a 800 nm. La fibrilla è costituita da catene polipeptidiche con conformazione cross-beta, che determina la birifrangenza quando colorata con il rosso Congo, caratteristico dell'amiloide. Oltre alla proteina fibrillare, l'amiloide contiene un'altra proteina, la cosiddetta componente P, che è la stessa in tutte le forme di amiloidosi. Il ruolo della componente P nell'amiloidogenesi non è chiaro. Potrebbe trattarsi di una normale proteina sierica che si lega alle fibrille amiloidi.
Virchow usò il termine botanico "amiloide" (dal greco amylon - amido) per descrivere il materiale extracellulare trovato nel fegato durante l'autopsia, poiché riteneva che fosse simile nella struttura all'amido. Successivamente è stata stabilita la natura proteica dei depositi, ma il termine “amiloide” è stato conservato fino ad oggi.
Classificazione dell'amiloidosi:
1. Amiloidosi primaria: non ci sono segni di malattia preesistente o coesistente; Tipo di deposito AL.
2. Amiloidosi combinata con mieloma: tipo AL.
3. Amiloidosi secondaria (reattiva): tipo AA; associati a infezioni croniche o malattie infiammatorie.
4. Tipo AF amiloidosi familiare ereditaria; l'amiloidosi è associata alla febbre mediterranea ereditaria (tipo AA) e a una serie di altre sindromi (tipo AF).
5. Amiloidosi locale.
6. Amiloidosi senile: associata all'invecchiamento; colpisce soprattutto il cuore e il cervello.
7. Amiloidosi associata ad emodialisi cronica.
Eziologia E patogenesi. L'eziologia dell'amiloidosi primaria è sconosciuta e la patogenesi dell'amiloidosi, così come le ragioni del danno predominante su alcuni organi nelle sue diverse varianti, rimangono non del tutto chiare. I fattori che contribuiscono allo sviluppo dell'amiloidosi comprendono la disproteinemia, principalmente l'iperglobulinemia, che riflette la funzione proteica-sintetica pervertita del sistema reticoloendoteliale, e i cambiamenti immunologici legati principalmente al sistema immunitario cellulare (soppressione del sistema T, cambiamenti nella fagocitosi, ecc.) . Apparentemente molti disturbi, in particolare i cambiamenti nelle reazioni cellulari, sono causati da caratteristiche genetiche, poiché vengono rilevati già nelle prime fasi della malattia, rimangono stabili durante la sua ulteriore progressione e sono standard in tutte le varianti dell'amiloidosi.
Esistono amiloidosi idiopatica (primaria), acquisita (secondaria), ereditaria (genetica), locale, amiloidosi nel mieloma e amiloidosi APUD.
Più comune amiloidosi secondaria, che in origine è vicina a reazioni aspecifiche (in particolare immunitarie). Si sviluppa con artrite reumatoide, spondilite anchilosante, tubercolosi, suppurazione cronica - osteomielite, bronchiectasie, meno spesso con linfogranulomatosi, tumori del rene, del polmone e di altri organi, sifilide, colite ulcerosa, morbo di Crohn e Whipple, endocardite infettiva subacuta, peoriasi, ecc. Casisticamente, l'amiloidosi si osserva raramente nelle malattie diffuse del tessuto connettivo, nella sarcoidosi.
Amiloidosi ereditaria nel nostro Paese è solitamente associata ad una malattia periodica, che si trasmette con modalità autosomica dominante. In questa malattia, l’amiloidosi può essere l’unica manifestazione. Altre forme di amiloidosi ereditaria sono meno comuni. In particolare, si distingue l'amiloidosi neuropatica portoghese, caratterizzata da polineuropatia periferica, compromissione della funzione intestinale, talvolta alterazioni della conduzione intracardiaca, impotenza.I depositi di amiloide si trovano in molti organi, principalmente nelle pareti dei piccoli vasi e dei nervi. Una variante simile dell'amiloidosi familiare è stata descritta in Giappone. Un'altra forma di amiloidosi neuropatica ereditaria che colpisce gli arti superiori, principalmente le mani, e opacità del vitreo è stata riscontrata in una famiglia di origine svizzera. Una variante simile di amiloidosi con un modello di trasmissione autosomica dominante è stata osservata in una famiglia tedesca. La variante finlandese è l'amiloidosi con atrofia corneale e neuropatia cranica.Sono note anche l'amiloidosi cardiopatica ereditaria (tipo danese), l'amiloidosi nefropatica con sordità, febbre e orticaria.
A differenza dell'amiloidosi secondaria ed ereditaria, l'amiloidosi primaria Non è possibile stabilire la causa o la natura ereditaria della malattia. In termini di struttura dell'amiloide e natura del danno agli organi interni, l'amiloidosi nel mieloma è vicina all'amiloidosi primaria, che è classificata in un gruppo separato. Recentemente, l'attenzione è stata prestata allo sviluppo dell'amiloidosi in età avanzata (specialmente nelle persone di età superiore ai 70-80 anni), quando sono colpiti il cervello, l'aorta, il cuore e il pancreas. Vengono descritte nuove forme di amiloidosi, in particolare l'amiloidosi in pazienti in emodialisi cronica, caratterizzata da artropatia distruttiva, sindrome del tunnel carpale e difetti ossei. La questione della relazione tra amiloidosi e aterosclerosi rimane aperta fino ad oggi, sebbene vi siano indicazioni che i cambiamenti ateromatosi possano contribuire alla deposizione di amiloide.
L'amiloidosi APUD è un tipo speciale di forma endocrina locale di amiloidosi, in cui la formazione del componente principale della fibrilla amiloide avviene dai prodotti di scarto delle cellule del sistema APUD (apudociti), tipico dei tumori - apudom. L'amiloidosi APUD comprende l'amiloidosi dello stroma del cancro midollare della tiroide, delle isole pancreatiche, dell'adenoma paratiroideo, dell'ipofisi e dell'amiloidosi atriale senile isolata.
Attualmente, l’amiloidosi è clinicamente divisa in forme sistemiche e locali.
Forme sistemiche di amiloidosi (classificazione in base alla composizione biochimica delle fibrille amiloidi):
1.tipo AL- composizione delle fibrille: catene leggere di immunoglobuline o loro frammenti (malattie associate: amiloidosi primaria, mieloma, malattia di Waldenström, linfomi maligni a cellule B);
2.tipo AA.- composizione delle fibrille: alfaglobulina SAA di fase acuta, simile nelle sue proprietà alla proteina C-reattiva (malattie associate: amiloidosi secondaria nelle malattie infiammatorie e reumatiche croniche, tumori, nonché nelle malattie periodiche, sindrome di Muckle-Wells);
3.tipo A-beta2M- composizione delle fibrille: 2-microglobulina (malattie associate: insufficienza renale cronica, si sviluppa a causa di una forte diminuzione dell'escrezione di questa proteina da parte dei reni e dell'impermeabilità delle membrane di dialisi ad essa);
4.tipo ATTR- composizione delle fibrille: proteina di trasporto transferrina - mutante e normale (malattie associate: con transferrina mutante - forme ereditarie familiari (portoghese, giapponese, ecc.), e con transferrina normale - forme senili*).
Forme locali di amiloidosi:
1 - Morbo di Alzheimer (A-beta, le fibrille sono costituite da proteine depositate nel cervello);
2 - amiloidosi delle isole pancreatiche (probabilmente avente una connessione patogenetica con il diabete di tipo 2);
3 - amiloidosi derivante da tumori endocrini;
4 - tumori amiloidi della pelle, della regione nasofaringea, della vescica e altri tipi rari.
* Amiloidosi senile. Sono state identificate forme sia sistemiche che locali di amiloidosi senile. L'amiloidosi sistemica senile è rara (dovrebbe essere differenziata dall'amiloidosi AL). Predominano le forme locali di amiloidosi senile, rappresentate sia dall'amiloidosi endocrina che da quella non endocrina. Le forme endocrine comprendono l'amiloidosi atriale senile isolata e l'amiloidosi senile delle isole pancreatiche, mentre le forme non endocrine comprendono l'amiloidosi aortica senile, l'amiloidosi cerebrale senile, l'amiloidosi oculare senile e l'amiloidosi senile della prostata e/o delle vescicole seminali. Oltre alle manifestazioni più comuni di un singolo organo dell'amiloidosi senile locale, sono possibili anche manifestazioni di più organi: l'amiloidosi senile multiorgano (combinata), che si riscontra in non più del 5% dei casi. Le più comuni sono le combinazioni di forme endocrine di amiloidosi con amiloidosi aortica o di amiloidosi delle isole pancreatiche con amiloidosi cerebrale e amiloidosi oculare associata. Le manifestazioni cliniche dell'amiloidosi senile sono estremamente diverse: disfunzione miocardica, diabete mellito non insulino-dipendente, demenza presenile e senile, cataratta, glaucoma, degenerazione maculare senile, ecc. Tuttavia, durante la vita, l'amiloidosi senile viene riconosciuta estremamente raramente.
Manifestazioni cliniche. I sintomi e il decorso dell’amiloidosi sono vari. Può includere proteinuria, sindrome nefrosica, azotemia, insufficienza cardiaca congestizia, cardiomegalia, aritmie, lesioni cutanee con papule cerose, ostruzione o ulcerazione gastrointestinale, sanguinamento, perdita di proteine, diarrea, macroglossia, dismotilità esofagea, neuropatia, ipotensione posturale, artrite, ostruzione respiratoria e deficit selettivo di fattori della coagulazione. Le manifestazioni cliniche dipendono dalla posizione dei depositi di amiloide, dal grado della loro prevalenza negli organi, dalla durata della malattia e dalla presenza di complicanze. Il quadro clinico diventa più complesso quando vengono colpiti i reni, la sede più comune, così come l'intestino, il cuore e il sistema nervoso. Il danno renale è tipico non solo nella forma secondaria più comune di amiloidosi, ma anche in quella primaria, inclusa l'amiloidosi ereditaria.
Diagnostica. La comparsa e la progressione della proteinuria, in particolare l'insorgenza di sindrome nefrosica o di insufficienza renale, in presenza di segni clinici o anamnestici di una malattia in cui può svilupparsi l'amiloidosi, è di fondamentale importanza per la diagnosi. Si dovrebbe pensare all'amiloidosi anche in assenza di indicazioni di tale malattia, soprattutto quando l'essenza della nefropatia rimane poco chiara o vi è insufficienza circolatoria, sindrome da malassorbimento e polineuropatia, che sono difficili da spiegare con altri motivi. Una diagnosi affidabile dell'amiloidosi richiede il rilevamento dell'amiloide nelle biopsie dei tessuti colpiti dopo colorazione speciale con rosso Congo (principalmente i reni, poi la mucosa del retto, meno spesso il tessuto gengivale). L'elettroforesi e l'immunoelettroforesi del siero del sangue e delle urine aiutano a determinare le paraproteine. L'aspirazione del tessuto adiposo addominale o la biopsia della mucosa rettale rivelano fibrille amiloidi. La conferma morfologica è necessaria in tutti i casi in cui uno studio accurato dell'anamnesi, compresa l'anamnesi familiare, le manifestazioni cliniche e i parametri di laboratorio rendono probabile la diagnosi di amiloidosi.
Previsione E trattamento. La prognosi varia e dipende dalla causa della malattia. La prognosi del mieloma è sfavorevole, ma la remissione è possibile per le infezioni croniche curabili. L’insufficienza renale è la principale causa di morte nell’amiloidosi avanzata. L’efficacia dei regimi di gestione dell’amiloidosi non è stata completamente dimostrata. Le raccomandazioni per il regime generale e la nutrizione per l'amiloidosi corrispondono a quelle per la nefrite cronica. È importante trattare attivamente la malattia che ha portato allo sviluppo dell'amiloidosi. Il volume della terapia sintomatica è determinato dalla gravità di alcune manifestazioni cliniche. Ai pazienti viene prescritto l'apporto di fegato crudo a lungo termine (8-10 mesi) (100-120 g/giorno). Nelle fasi iniziali della malattia vengono prescritti farmaci a base di 4-aminochinolina, ad esempio delagil 0,25 g una volta al giorno, sotto monitoraggio a lungo termine del numero di leucociti nel sangue (possibile sviluppo di leucopenia) e dei mezzi di rifrazione del occhi (possibilità di deposizione di derivati farmaceutici). Vengono utilizzati anche Unithiol (6-10 ml di una soluzione al 5% al giorno per via intramuscolare in cicli ripetuti di 30 giorni con interruzioni di 1-2 mesi), dimetilsolfossido (5-8 g di farmaco al giorno per molti mesi) e tentativi sono fatti per usare la colchicina. La colchicina (1 - 2 mg al giorno) può bloccare la deposizione di amiloide. L'amiloidosi primaria può rispondere al trattamento con prednisolone con un agente alchilante (melfolan), probabilmente perché questi farmaci agiscono prendendo di mira la sintesi della proteina amiloide AL. Alcuni pazienti sono indicati per il trapianto di rene.
Regimi di trattamento per l'amiloidosi primaria. (1) Somministrazione orale ciclica di melfolan (0,15-0,25 mg/kg di peso corporeo al giorno) e prednisolone (1,5-2,0 mg/kg al giorno) per quattro-sette giorni ogni quattro-sei settimane per anni, fino alla dose di ciclo di Si raggiungono i 600 mg. (2) Uso orale di melfolan alla dose di 4 mg/giorno per tre settimane, quindi, dopo una pausa di due settimane, 2-4 mg/giorno quattro giorni alla settimana ininterrottamente, fino al raggiungimento della dose di 600 mg, in combinazione con prednisolone. (3) Somministrazione endovenosa di alte dosi di melfolan (100-200 mg/m² di superficie corporea per due giorni) seguita da trapianto autologo di cellule staminali. (4) Desametasone endovenoso 40 mg per quattro giorni ogni tre settimane per otto cicli. (5) Somministrazione endovenosa di desametasone alla dose di 40 mg nel primo quarto, 9-12 e 17-20 giorno di un ciclo di 35 giorni, da tre a sei cicli, seguito dall'uso di interferone α alla dose di 3-6 milioni di unità tre volte a settimana. (6) Regime vincristina-doxoribucina-desametasone (VAD).
L'amiloidosi è una malattia sistemica in cui l'amiloide (una sostanza proteica-polisaccaridica (glicoproteina)) si deposita negli organi e nei tessuti, provocando l'interruzione delle loro funzioni.
L'amiloide è costituita da proteine globulari e fibrillari strettamente intrecciate con i polisaccaridi. Una lieve deposizione di amiloide nei tessuti ghiandolari, nello stroma degli organi parenchimali e nelle pareti dei vasi sanguigni non causa alcun sintomo clinico. Ma con depositi significativi di amiloide negli organi, si verificano cambiamenti macroscopici pronunciati. Il volume dell'organo interessato aumenta, i suoi tessuti acquisiscono una lucentezza cerosa o grassa. Successivamente, si sviluppa l'atrofia degli organi con la formazione di insufficienza funzionale.
L’incidenza dell’amiloidosi è di 1 su 50.000 persone. La malattia è più comune nelle persone anziane.
La deposizione di amiloide è un segno di amiloidosi
Cause e fattori di rischio
L'amiloidosi di solito si sviluppa sullo sfondo di malattie infiammatorie purulente a lungo termine (endocardite batterica, bronchiectasie, osteomielite) o malattie infettive croniche (malaria, actinomicosi, tubercolosi). L'amiloidosi si sviluppa un po' meno frequentemente nei pazienti affetti da cancro:
- cancro ai polmoni;
- cancro al rene;
- leucemia;
- linfogranulomatosi.
L'amiloidosi può colpire vari organi e il quadro clinico della malattia è vario.
Inoltre, le seguenti malattie possono portare all’amiloidosi:
- sarcoidosi;
- la malattia di Whipple;
- Morbo di Crohn;
- colite ulcerosa non specifica;
- psoriasi;
- La malattia di Bekhterev;
- artrite reumatoide;
- aterosclerosi.
Non esistono solo forme acquisite, ma anche ereditarie di amiloidosi. Questi includono:
- febbre mediterranea;
- Amiloidosi neuropatica portoghese;
- amiloidosi finlandese;
- Amiloidosi danese.
Fattori che causano l’amiloidosi:
- predisposizione genetica;
- violazioni dell'immunità cellulare;
- iperglobulinemia.
Forme della malattia
A seconda dei motivi che l’hanno provocata, l’amiloidosi si divide in diverse forme cliniche:
- senile (senile);
- ereditario (genetico, familiare);
- secondario (acquisito, reattivo);
- idiopatico (primario).
A seconda dell'organo in cui vengono depositati prevalentemente i depositi di amiloide, si distinguono:
- amiloidosi dei reni (forma nefrotica);
- amiloidosi cardiaca (forma cardiopatica);
- amiloidosi del sistema nervoso (forma neuropatica);
- amiloidosi epatica (forma epatica);
- amiloidosi surrenale (forma epinefropatica);
- ARUD-amiloidosi (amiloidosi degli organi del sistema neuroendocrino);
- amiloidosi mista.

L’amiloidosi può anche essere locale o sistemica. Con l'amiloidosi locale, c'è una lesione predominante di un organo, con l'amiloidosi sistemica - due o più.
Sintomi
Il quadro clinico dell'amiloidosi è vario: i sintomi sono determinati dalla durata della malattia, dalla localizzazione dei depositi di amiloide e dalla loro intensità, dal grado di disfunzione d'organo e dalle peculiarità della struttura biochimica dell'amiloide.
Nella fase iniziale (latente) dell’amiloidosi non ci sono sintomi. La presenza di depositi di amiloide può essere rilevata solo al microscopio. Successivamente, con l'aumento dei depositi della glicoproteina patologica, si verifica e progredisce il fallimento funzionale dell'organo interessato, che determina le caratteristiche del quadro clinico della malattia.
Fattori che causano l'amiloidosi: predisposizione genetica, compromissione dell'immunità cellulare, iperglobulinemia.
Con l'amiloidosi renale, si osserva una proteinuria moderata per lungo tempo. Quindi si sviluppa la sindrome nefrosica. I principali sintomi dell’amiloidosi renale sono:
- la presenza di proteine nelle urine;
- ipertensione arteriosa;
- rigonfiamento;
- fallimento renale cronico.
L’amiloidosi cardiaca è caratterizzata da una triade di sintomi:
- disturbo del ritmo cardiaco;
- cardiomegalia;
- insufficienza cardiaca cronica progressiva.
Nelle fasi successive della malattia, anche uno sforzo fisico minore porta a grave debolezza e mancanza di respiro. Sullo sfondo dell'insufficienza cardiaca, la polisierosite può svilupparsi:
- pericardite da versamento;
- pleurite da versamento;
- ascite.
Con l'amiloidosi del tratto gastrointestinale, l'attenzione è attirata dall'allargamento della lingua (macroglossia), che è associato alla deposizione di amiloide nello spessore del suo tessuto. Altre manifestazioni:
- nausea;
- bruciore di stomaco;
- stitichezza, seguita da diarrea;
- alterato assorbimento dei nutrienti dall'intestino tenue (sindrome da malassorbimento);
- sanguinamento gastrointestinale.
Le lesioni amiloidi del pancreas di solito si manifestano sotto le sembianze di pancreatite cronica. La deposizione di amiloide nel fegato causa ipertensione portale, colestasi ed epatomegalia.
Con l'amiloidosi della pelle, compaiono noduli cerosi nel collo, nel viso e nelle pieghe naturali. Spesso l'amiloidosi cutanea nel suo decorso ricorda il lichen planus, la neurodermite o la sclerodermia.

Con l'amiloidosi del sistema muscolo-scheletrico, il paziente sviluppa:
- miopatia;
- periartrite omeroscapolare;
- sindrome del tunnel carpale;
- poliartrite che colpisce le articolazioni simmetriche.
L’amiloidosi del sistema nervoso è grave ed è caratterizzata da:
- mal di testa persistente;
- vertigini;
- demenza;
- aumento della sudorazione;
- collasso ortostatico;
- paralisi o paresi degli arti inferiori;
- polineuropatia.
Diagnostica
Tenendo conto del fatto che l'amiloidosi può colpire vari organi e che il quadro clinico della malattia è vario, la sua diagnosi presenta alcune difficoltà. Lo stato funzionale degli organi interni può essere valutato mediante:
- EcoCG;
- radiografia;
- gastroscopia (EGD);
- sigmoidoscopia.
L'incidenza dell'amiloidosi è di 1 caso ogni 50.000 persone. La malattia è più comune nelle persone anziane.
L'amiloidosi può essere sospettata se nei risultati dei test di laboratorio vengono rilevati i seguenti cambiamenti:
- anemia;
- trombocitopenia;
- ipocalcemia;
- iponatriemia;
- iperlipidemia;
- ipoproteinemia;
- cilindriruria;
- leucocituria.
Per la diagnosi definitiva è necessario eseguire una biopsia puntura dei tessuti interessati (mucosa del retto, stomaco, linfonodi; gengive; reni) seguita dall'esame istologico del materiale ottenuto. Il rilevamento di fibrille amiloidi nel campione in esame confermerà la diagnosi.
Trattamento
Nel trattamento dell'amiloidosi primaria vengono utilizzati ormoni glucocorticoidi e farmaci citostatici.
Nell’amiloidosi secondaria, il trattamento è mirato principalmente alla malattia di base. Vengono prescritti anche medicinali della serie 4-aminochinolina. Si consiglia una dieta a basso contenuto proteico con sale limitato.
Lo sviluppo di insufficienza renale cronica allo stadio terminale è un'indicazione all'emodialisi.

Possibili complicazioni e conseguenze
L'amiloidosi può essere complicata dalle seguenti patologie:
- diabete;
- insufficienza epatica;
- sanguinamento gastrointestinale;
- insufficienza renale;
- ulcere amiloidi dello stomaco e dell'esofago;
- insufficienza cardiaca.
Previsione
L’amiloidosi è una malattia cronica e progressiva. Nell'amiloidosi secondaria, la prognosi è in gran parte determinata dalla possibilità di trattare la malattia di base.
Man mano che si sviluppano complicazioni, la prognosi peggiora. Una volta che compaiono i sintomi dell’insufficienza cardiaca, l’aspettativa di vita media è solitamente inferiore a pochi mesi. L’aspettativa di vita dei pazienti con insufficienza renale cronica è in media di 12 mesi. Questo periodo aumenta leggermente in caso di emodialisi.
Prevenzione
Non esiste alcuna prevenzione per l’amiloidosi primaria (idiopatica), poiché la causa è sconosciuta.
Per prevenire l’amiloidosi secondaria è importante identificare e trattare tempestivamente le malattie infettive, oncologiche e infiammatorie purulente.
La prevenzione delle forme genetiche di amiloidosi consiste nella consulenza medica e genetica delle coppie sposate nella fase di pianificazione della gravidanza.
Video da YouTube sull'argomento dell'articolo:
Amiloide, sua natura e proprietà.
Nel 1853, Virchow dimostrò che le sostanze depositate nei tessuti durante la "malattia grassa" di Rokitansky hanno la proprietà di macchiarsi di iodio, come l'amido, e la chiamò amiloide.
Composizione chimica e proprietà fisiche dell'amiloide.
L'amiloide è una sostanza complessa - una glicoproteina, in cui le proteine fibrillare e globulari sono associate ai polisaccaridi.
Proteine amiloidi .
Secondo Kravkov (1894), l'amiloide comprende:
Carbonio (48,86 - 50,38%);
Idrogeno (6,45 - 7,09%);
Azoto (13,79 - 14,67%);
Zolfo (2,65 - 2,8%);
Fosforo (tracce).
Giles e Calkins (1955) trovarono nell'amiloide l'82,8% di acqua, fino al 14,2% di azoto, fino al 4% di carboidrati, 0,9% di fosforo e 0,86% di zolfo.
L'amiloide produce le reazioni del biureto e della xantoproteina caratteristiche delle proteine. Un'alta percentuale di contenuto di acqua nell'amiloide indica l'elevata idrofilicità delle proteine, e un basso contenuto di fosforo suggerisce che appartiene agli acidi nucleici presenti nell'amiloide, sebbene non ci siano dati affidabili). L'amiloide è pironinofila.
La composizione aminoacidica dell'amiloide differisce dalle proteine del siero e dei tessuti (in percentuali molari):
Glicina - 10-17,3;
Alanina - 9,7-14,4;
Leucina - 7,7-13,7;
Valina - 4,1 - 7,1;
Metionina - 1,1;
Cisteina - 1,5 - 2,9;
Tirosina - 2,7-3,7;
Triptofano;
Istidina.
L'amiloide è dominata da gruppi lipofili (glicina, alanina, leucina, valina).
La componente proteica dell'amiloide è simile alle globuline. Il contenuto di metionina e cisteina si è rivelato vicino a quello ottenuto dall'analisi delle gammaglobuline sieriche, sebbene il contenuto di aminoacidi nell'amiloide e nelle gammaglobuline sieriche sia diverso.
La presenza di triptofano indica la sua vicinanza al fibrinogeno, che contiene anch'esso questo amminoacido in grandi quantità. L'amiloide contiene molta tirosina (ialina e collagene). Qualitativamente, la composizione aminoacidica dell'amiloide è la stessa nelle diverse forme di amiloidosi, tuttavia, le caratteristiche quantitative dei componenti inclusi nella sua composizione variano a seconda non solo della forma di amiloidosi, ma anche dell'organo in cui l'amiloide è depositata .
Sulla base di un'analisi della composizione aminoacidica, Letterer (1955) concluse che l'amiloide è una miscela di due proteine: una di queste è vicina alle globuline sieriche, l'altra al collagene. Le proteine amiloidi sono costituite da due frazioni con proprietà diverse. La frazione A (85-90%) è insolubile in acqua, precipitata con acido acetico, dopo precipitazione in tampone acetato o fosfato a pH 3,9 - 6,4, elettroforeticamente vicina alle alfa-1 e alle gamma globuline sieriche e ha proprietà antigeniche. La frazione B (10-15%) è solubile in acqua, precipitata dall'alcool e ha una mobilità elettroforetica vicina a quella delle beta globuline.
Le proteine plasmatiche nella composizione dell'amiloide sono considerate "additivi", il cui aspetto è spiegato dall'adsorbimento non specifico di molte sostanze nell'amiloide a causa delle peculiarità della sua fine struttura fibrillare.
Carboidrati amiloidi.
I polisaccaridi costituiscono il 2-4% della massa totale dell'amiloide e sono rappresentati da una quantità equivalente di glucosio, galattosio, una quantità leggermente inferiore di galattosamina ed esosamina, mannosio e fruttosio. I carboidrati sono costituiti da due frazioni: neutra e acida.
1. La prima frazione, PAS - positiva, non dà metacromasia, ha mobilità elettroforetica come le alfa-2 e le gamma globuline nel siero. È classificata come glucoproteina sierica, il che è confermato dall'alto contenuto di esosamina e acido neuraminico nell'amiloide e nel siero del sangue durante l'amiloidosi, che fanno parte dei gruppi di carboidrati associati alle globuline sieriche.
2. La seconda frazione è metacromatica, la base dei polisaccaridi acidi è condroitin solfato (90%), eparitin solfato, acido ialuronico, condroitina ed eparina. I mucopolisaccaridi non sono di origine ematogena, ma del tessuto connettivo locale, e l'aumento di MPS avviene senza disturbare il rapporto tra condroitin solfato ed eparitin solfato insito nel tessuto normale. Secondo Muir e Cohen (1968), oltre al condroitin solfato (50%) e all'eparitin solfato (30%), è incluso il cheratina solfato (20%).
La frazione A è associata ai polisaccaridi sierici PAS-positivi, la frazione B è associata ai mucopolisaccaridi acidi (con il blu di toluidina dà metacromasia).
Oltre a proteine e carboidrati, lipidi e lipoproteine, nell'amiloide sono stati trovati sali di calcio (integratori di amiloide).
Proprietà fisiche dell'amiloide.
Tra le proprietà fisiche dell'amiloide, sono caratteristici l'anisotropia positiva e il dicroismo. Se colorato con rosso Congo, la birifrangenza aumenta. L'anisotropia dell'amiloide indica la struttura molecolare ordinata della sua sostanza. Basandosi sulle proprietà ottiche di polarizzazione dell'amiloide, Romhanyi ritiene che abbia una struttura paracristallina submicroscopica fibrillare fine.
La forza dei legami proteina-polisaccaride spiega la resistenza dell'amiloide all'azione degli enzimi (ad esempio: la tripsina porta ad un indebolimento della reazione PAS dell'amiloide, ma non modifica la colorazione ortocromatica e la birifrangenza).
La colorazione dell'amiloide con il rosso Congo è determinata dalle caratteristiche conformazionali delle proteine e delle sue fibrille.
Componenti ultrastrutturali.
Nell'amiloide sono stati trovati due componenti ultrastrutturali: fibrille e bastoncini periodici. Le fibrille hanno un diametro di 7,5 nm e una lunghezza fino a 800 nm. Ciascuna fibrilla è composta da due subfibrille con un diametro di 2,5 nm, che si trovano parallele ad una distanza di 2,5 nm. Le fibrille sono striate e nastriformi. Secondo Wolman (1971) fibrille parallele di proteine e polisaccaridi amiloidi neutri sono intrecciate con fibrille di mucopolisaccaridi acidi. La proteina amiloide fibrillare (componente F) è caratterizzata da un alto contenuto di triptofano, acido dicarbossilico, catene corte di aminoacidi e un basso contenuto di idrossiprolina, idrossilisina: questo distingue l'amiloide dal collagene, dalla reticolina, dall'elastina. Le fibrille rappresentano un gruppo eterogeneo di proteine con caratteristiche individuali; in ogni caso di amiloidosi, la composizione aminoacidica della proteina fibrillare è diversa. Da una miscela di proteine della fibrilla amiloide vengono isolate la proteina A (AS) e la proteina B. La proteina A (AS), rispetto alla proteina B, contiene più aminoacidi come arginina, asparagina, glicina, alanina e fenilalanina; ha una struttura antigenica specifica. La proteina B, presente nell'amiloide nell'amiloidosi primaria, l'amiloidosi tumorale, ha un grande peso molecolare relativo, simile nella composizione aminoacidica alle catene leggere delle immunoglobuline. Le proteine amiloidi sono caratterizzate da un ripiegamento delle catene polipeptidiche (conformazione cross-beta).
I bastoncini periodici costituiscono il 5% rispetto alle fibrille. Il loro diametro è di 10 nm, la lunghezza fino a 250 nm. Sono costituiti da formazioni peptagonali con un diametro di 9-10 nm e una larghezza di 2 nm, situate a una distanza di 4 nm l'una dall'altra. Differiscono dalle fibrille non solo nella composizione quantitativa degli aminoacidi (predominano glicina, leucina, glutammina e asparagina, il triptofano è assente), ma anche nella composizione dei carboidrati (basso contenuto di esosamina e acido uronico, alto contenuto di acido neuraminico e esoso). Rispetto alle proteine delle fibrille, i bastoncini periodici sono un antigene più forte. I bastoncini periodici (componente P) corrispondono all'alfa globulina plasmatica.
Patogenesi dell'amiloidosi.
Molte teorie sono state proposte per spiegare la patogenesi dell’amiloidosi. Ad esempio, Gzerny (1893) e Shepilevsky (1899) consideravano l'amiloide un prodotto dell'infiammazione.
N. P. Kravkov (1898) credeva che l'amiloide si formasse come risultato dell'attività vitale dei microrganismi; secondo A. A. Maksimov (1896), la sostanza amiloide appare come risultato della disorganizzazione dei tessuti. Finora, 3 teorie sulla patogenesi dell'amiloidosi non hanno perso il loro significato: la teoria della disproteinosi, la teoria immunologica e la teoria della secrezione cellulare e locale.
La teoria della disproteinosi.
La creazione di questa teoria è associata al nome di Virchow, che considerava l'amiloide un "prodotto del sangue". Questa teoria considera l'amiloide come un prodotto del metabolismo proteico compromesso. Il collegamento principale nella patogenesi è la disproteinemia con l'accumulo nel plasma di frazioni proteiche grossolane e proteine anomale (paraproteine) che, dopo aver oltrepassato il letto vascolare, formano una sostanza amiloide.
È noto che lo sviluppo dell'amiloidosi è preceduto da uno stato di disproteinemia (iperglobulinemia, iperfibrinogenemia, ipoalbuminemia, paraproteinemia, ecc.). Si osserva nelle malattie che portano all'amiloidosi secondaria (tubercolosi, artrite reumatoide, osteomielite cronica, linfogranulomatosi, plasmocitoma), nelle malattie periodiche e nell'amiloidosi idiopatica.
L'amiloide nel plasmocitoma è costituita da catene leggere dell'amminoacido. È stata stabilita la somiglianza della composizione e della sequenza aminoacidica della proteina della fibrilla amiloide (proteina B) nel plasmocitoma con i frammenti N-terminali delle catene leggere delle immunoglobuline.
La disproteinemia caratterizza lo stadio pre-amiloide. Ciò avviene con l'introduzione di microrganismi, tossine, proteine ed enzimi proteolitici. In questa fase progredisce l'aumento delle gamma globuline nel sangue, preceduto da un aumento delle globuline alfa e beta e delle corrispondenti glucoproteine. Si verifica la formazione di proteine anomale assenti nel sangue degli animali di controllo.
Jaeger (1960) notò un calo del livello di gammaglobuline sieriche anomale con la comparsa di sostanza amiloide nei tessuti, collegando questo fatto con il consumo di proteine anomale per la costruzione dell'amiloide.
La cessazione delle iniezioni di amiloidogeno ha portato alla normalizzazione dei livelli sierici di beta-globuline e ad un aumento delle gamma-globuline, accompagnato dal riassorbimento dell'amiloide nella milza e nei reni.
Un collegamento essenziale nella patogenesi dell'amiloidosi, che viene spiegata con la teoria della disproteinosi, è il rilascio di proteine (paraproteine) circolanti nel plasma nei tessuti. L'amiloide si forma sotto l'endotelio e la membrana argirofila del vaso, che indica la permeabilità della parete vascolare alle paraproteine.
I processi di polimerizzazione e depolimerizzazione degli MPS acidi della sostanza principale sono più facilmente realizzabili e più facilmente distrutti vicino alla barriera endoteliale, dove vengono eseguiti sotto l'influenza delle ialuronidasi plasmatiche e tissutali. La deposizione subendoteliale di amiloide riflette i cambiamenti nel sistema enzimatico (ialuronidasi), portando all'interruzione dei ritmi fisiologici di polimerizzazione e depolimerizzazione: ciò determina la possibilità di combinare paraproteine e glucoproteine plasmatiche con proteine fibrillari e MPS della sostanza principale, che porta alla formazione di amiloide.
La localizzazione subendoteliale dell'amiloide supporta una teoria che sottolinea il significato primario dei disturbi nella sintesi delle proteine corporee; essa spiega la localizzazione selettiva dell'amiloide negli organi. Dietro la barriera endoteliale (negli organi di escrezione e immagazzinamento) si accumulano prodotti della sintesi proteica ridotta rilasciati dal sangue, che è spesso associata all'amiloidosi dei reni, del fegato, dell'intestino e della milza. Tuttavia, questa teoria è troppo semplice nelle sue disposizioni e necessita pertanto di modifiche.
La disproteinemia non è specifica per l'amiloidosi ed è espressione di un metabolismo proteico compromesso, caratteristico di processi cronici purulento-distruttivi, iperimmunizzazione, autoimmunizzazione e malattie metaboliche ereditarie. La teoria della disproteinosi non tiene conto del ruolo delle trasformazioni cellulari del RES e non rivela il meccanismo di sviluppo della disproteinemia stessa. Spiega il meccanismo di formazione di una specifica proteina amiloide nel plasmocitoma e non è in grado di rispondere dove e come si formano le proteine e le glucoproteine, che sono il “materiale da costruzione” della sostanza amiloide.
Teoria immunologica.
I fondatori di questa teoria, che considera la formazione dell'amiloide come risultato della reazione antigene-anticorpo, sono considerati Loeschke (1927) e Letterter (1934). Sono partiti dal fatto che nelle malattie complicate dall'amiloidosi si formano prodotti di degradazione dei tessuti, leucociti e tossine batteriche, che possono avere proprietà antigeniche e portare alla produzione di anticorpi. La formazione di amiloide è il risultato della reazione di precipitazione di un complesso proteico nei siti di produzione di anticorpi (APE). Letterer trovò precipitine specifiche per le proteine tissutali nel sangue di animali affetti da amiloidosi, dimostrando così la loro natura autoimmune, e stabilì che l'amiloidosi si verifica in condizioni di scarsa produzione di anticorpi, con il loro contenuto insufficiente nel sangue e nei tessuti e una quantità eccessiva di antigeni. Pertanto, la frequenza dell'amiloidosi durante i processi suppurativi indica indirettamente l'importanza della disgregazione dei tessuti e della disgregazione dei leucociti e lo sviluppo di una reazione autoimmune nell'amiloidogenesi.
Studi di Janigan e Druet (1966) hanno dimostrato che la capacità amiloidogenica di varie sostanze proteiche solubili aumenta parallelamente all'aumento delle loro proprietà antigeniche. L'acquisizione delle proprietà antigeniche è accompagnata dalla comparsa della capacità di provocare amiloidosi; con la perdita di antigenicità, la sostanza perde questa capacità.
Mellors e Ortega (1956) hanno dimostrato che l'amiloide può legarsi al siero omologo di antigammaglobuline in modo antigene-anticorpo; contiene anticorpi specifici contro l'antigene amiloidogeno e la localizzazione di anticorpi specifici coincide con la fissazione di gammaglobuline omologhe.
Negli studi istosierologici, Letterter e Lachmann (1969) trovarono il complemento nell'amiloide e, quindi, mostrarono la partecipazione della reazione antigene-anticorpo alla formazione dell'amiloide. Successivamente, nelle masse amiloidi sono stati trovati complessi antigene-anticorpo che fissano il complemento.
Nel 1957 Parnes V.A. hanno trovato complessi antigenici specifici (autoantigeni) nella milza amiloide di pazienti deceduti a causa di varie malattie. Lei ritiene che la natura autoantigenica dell'amiloidosi determini lo sviluppo di processi di autoimmunizzazione. Pavlikhina L.V. e Serov V.V. utilizzando la reazione di fissazione del complemento, hanno mostrato il graduale accumulo di anticorpi anti-organo nell'amiloidosi. Negli stadi successivi dell’amiloidosi, il titolo degli anticorpi anti-organo aumentava. L'amiloide ha una certa specificità come autoantigene.
Zubzhitsky Yu.N. (1967) scoprirono che nell'amiloidosi sperimentale dei topi, il complesso antigene estraneo-anticorpo omologo viene rilevato negli organi prima che vengano rilevati i depositi di amiloide. In questo caso, la distruzione dei tessuti, accompagnata dalla formazione di amiloide, è indotta dall'azione del complesso antigene-anticorpo in condizioni di eccesso di antigene estraneo. La comparsa degli anticorpi anti-organo riscontrati nel siero dei topi avviene dopo l'inizio della distruzione dei tessuti durante la formazione dell'amiloide, il che rende possibile riconoscere gli autoantigeni di questi tessuti come antigeni autoimmunizzati.
La fase di formazione del complesso autoimmune, comprendente l'antigene della caseina (anticorpo 7S), viene sostituita dalla fase di formazione della sostanza amiloide, che comprende una sostanza simile all'anticorpo. Questa sostanza presenta le proprietà degli anticorpi 19S e appare dopo l '"esaurimento" della capacità delle plasmacellule di produrre immunoglobuline normali.
Il ruolo principale nello sviluppo dell'amiloide è giocato dal fallimento dell'iperimmunizzazione: una diminuzione della funzione del sistema immunocompetente, la stimolazione delle trasformazioni cellulari che portano alla comparsa di cellule (amiloidoblasti) che secernono una specifica proteina amiloide fibrillare.
Rokosuev V.S. (1975) ritengono che qualsiasi stimolazione antigenica a lungo termine del corpo sia accompagnata dalla produzione di alcuni fattori non specifici ("fattore non specifico che stimola l'amiloidogenesi", "fattore di trasferimento") che realizzano lo sviluppo dell'amiloidosi. È stato stabilito che il "fattore di trasferimento", che sembra essere nelle cellule del tessuto linfoide, stimola la produzione di amiloide nei topi con un numero insufficiente di iniezioni dell'immunogeno quando viene trasferito da topi e persone con amiloidosi. Il "fattore di trasferimento" è di natura nucleoproteica. È stato dimostrato che la frazione nucleare grezza estratta dagli omogenati di milza di topi amiloidi causava l'induzione dell'amiloidosi. Cioè, durante l'amiloidosi avviene un trasferimento di informazioni da parte delle cellule del sistema linfoide; viene trasportato da uno specifico trasportatore immunitario RNA.
Si presume che l'amiloide sia il risultato dell'interazione di un fattore stimolante l'amiloide e di un antigene specifico. In questo caso, il ruolo delle cellule (amiloidoblasti) che producono amiloide sotto l'influenza di un fattore produttore di amiloide in presenza di antigeni specifici diventa eccezionale.
Con l’amiloidosi, l’immunità cellulare viene soppressa man mano che il processo progredisce.
Pertanto, la teoria immunologica dell’amiloidosi ha chiarito e integrato le idee sul ruolo delle reazioni immunitarie umorali nella formazione della sostanza amiloide. L’affermazione che “l’amiloide è un complesso antigene-anticorpo” non può essere accettata, ma ciò non significa che gli immunocomplessi non possano far parte dell’amiloide come “additivi” ematogeni. La presenza di immunocomplessi con amiloide indica il ruolo della risposta immunitaria umorale nella formazione dell'amiloide.
Nel 1973 Hardt citò i seguenti fatti: una forte stimolazione antigenica porta all'amiloidosi; le cellule “occupate” nelle fasi pre-amiloide e amiloide sono conosciute come “mediatori delle reazioni immunitarie”; nella fase amiloide si osserva la disfunzione delle cellule immunocompetenti e questi disturbi sono correlati ai cambiamenti negli organi linfoidi; le fibrille amiloidi sono costituite da componenti immunoglobulinici. La stimolazione antigenica a lungo termine porta alla disintegrazione dei linfociti T con il rilascio di sostanze di natura nucleoproteica che, insieme ad antigeni specifici, causano la produzione di fibrille amiloidi da parte delle cellule mesenchimali, cioè l'immunità umorale e cellulare viene interrotta. Questi disturbi determinano l'impoverimento del tessuto linfoide e la comparsa di un pool di cellule (amiloidoblasti) capaci di secernere una proteina fibrillare anomala, che, combinandosi nei tessuti con altre proteine e polisaccaridi, forma una glicoproteina complessa - amiloide.
La teoria immunologica rivela un collegamento importante nella patogenesi dell'amiloidosi: un cambiamento nell'omeostasi immunitaria. Ci consente di spiegare l'amiloidosi sperimentale e l'amiloidosi secondaria negli esseri umani (esclusa la paraamiloidosi), ma non è applicabile all'amiloidosi idiopatica e genetica.
Teoria della mutazione.
Questa teoria della formazione di un clone di cellule amiloidoblastiche può diventare universale nello spiegare la patogenesi di tutte le forme conosciute di amiloidosi, se immaginiamo la possibile varietà di fattori mutageni.
Nell'amiloidosi secondaria (esclusa l'amiloidosi nel plasmocitoma), le mutazioni e la comparsa di un clone di amiloidoblasto sono associati ad una stimolazione antigenica prolungata.
Nell'amiloidosi genetica (familiare), stiamo parlando di una mutazione genetica che si verifica in diversi loci, che determina la differenza nella composizione della proteina amiloide in diverse persone e animali. Nell'amiloidosi senile si verificano meccanismi simili, poiché l'amiloidosi senile è considerata una fenocopia di quella genetica. Le mutazioni cellulari nella para-amiloidosi e, possibilmente, nell'amiloidosi locale simile a un tumore, sono causate da mutageni tumorali. Poiché i precursori dell'amiloide - le fibrille amiloidi (le loro proteine) sono immunogeni deboli, le cellule mutate non vengono riconosciute dal sistema immunocompetente e non vengono eliminate. Si verifica tolleranza immunologica alle proteine amiloidi.
La teoria della mutazione dell'amiloidogenesi ha permesso di comprendere la vicinanza dell'amiloidosi al processo tumorale.
Parlando della relazione specifica tra amiloidosi e tumore, si distinguono tre varianti di combinazioni:
1. L’amiloidosi “complica” un tumore, solitamente un cancro di una sede o di un’altra. Si sviluppa un'amiloidosi secondaria e il meccanismo dell'amiloidogenesi è identico a quello della tubercolosi e della suppurazione cronica.
Link principali:
a) degradazione dei complessi antigene-anticorpo da parte dei macrofagi, attivazione da parte di prodotti di degradazione di cellule pironinofile e PAS-positive e sintesi dell'amiloide da parte degli amiloidoblasti utilizzando un “fattore di trasferimento” rilasciato dalle cellule pironinofile.
2. Amiloidosi nella reticolosi (plasmocitoma, reticolosarcoma, linfogranulomatosi, malattia di Waldenström).
Si sviluppa un'amiloidosi generalizzata, ma il suo sviluppo in questi casi è associato alla proliferazione tumorale di cellule come reticolari, plasmacellule e linfociti. Allo stesso tempo, ci sono fatti che l'amiloide nell'emoblastosi (paraproteinemica) è direttamente o indirettamente un prodotto delle cellule tumorali. Normalmente, gli elementi della linea "cellule reticolari - plasmacellule" sintetizzano le immunoglobuline e, nel processo di trasformazione del tumore, le proteine del mieloma, più spesso le catene L delle immunoglobuline, da cui viene costruita l'amiloide. Si suggerisce che le catene leggere delle immunoglobuline vengano catturate dai macrofagi, che processano le catene L e costruiscono amiloide da questi frammenti. Le catene L sono varianti filogeneticamente più primitive dei “precursori delle immunoglobuline”.
3. Amiloidosi dello stroma di tumori benigni o maligni. Più spesso si tratta di tumori degli organi endocrini o rappresentanti del sistema paracrino: cancro midollare della tiroide, insulinoma, carcinoidi, tumori della pelle e odontogeni - tumore di Teredberg. In questo caso, le cellule tumorali epiteliali partecipano alla formazione dell'amiloide. Nel cancro midollare della tiroide, l’amiloide è prodotta dalle cellule C parafollicolari, che normalmente producono calcitonina. Negli insulinomi, le cellule B producono amiloide o insulina, la cui molecola proteica, come l'amiloide e la cheratina, è caratterizzata da un'abbondanza di legami disolfuro. Apparentemente, l’amiloidosi senile delle isole di Langerhans (un componente della triade di Schwarz) è associata a questa produzione “errata” di amiloide da parte delle cellule B invece che dell’insulina, e l’amiloidosi cutanea locale è associata alla produzione di amiloide da parte delle cellule epidermiche invece dell’insulina. cheratina.
La connessione tra una cellula tumorale e l'amiloide nei tumori polipeptidici endocrini ("apudomi"), che normalmente producono polipeptidi e, come risultato della trasformazione del tumore, insieme ad altri segni di progressione del tumore, iniziano a sintetizzare una proteina anormale - "apud-amiloide ".
Pertanto, l'amiloide è il prodotto di una cellula che ha subito l'evoluzione del tumore.
Esistono molte somiglianze tra amiloidogenesi e oncogenesi. Pertanto, un segno di progressione dell'amiloidosi è rappresentato dalla proliferazione incontrollata di cellule RPE indifferenziate. La produzione progressiva incontrollata di proteina amiloide anormale da parte di cellule proliferanti di un tipo speciale può essere considerata un segno secondario di progressione (anaplasia), che si sviluppa autonomamente. Si presuppone che le cellule capaci di evoluzione tumorale siano un “pool” di cellule dormienti, disposte primitivamente, soggette a mutazioni somatiche (cellule che sintetizzano le fibrille amiloidi): si tratta degli amiloidoblasti, che sono derivati di elementi cellulari cambiali di origine mesenchimale.
La relazione tra amiloidosi e tumori dal punto di vista dei disturbi dell'equilibrio enzimatico della cellula (enzimopatia). L'azione del fattore oncogeno (virus, mutazione somatica) sembra essere basata sulla fermentopatia, principalmente RNA). Se intendiamo il processo di “disdifferenziazione” e di evoluzione tumorale delle cellule come un'enzimopatia causata dalla distruzione degli enzimi associati al metabolismo degli acidi nucleici, allora la “disdifferenziazione” delle cellule con la formazione di amiloidoblasti e la sintesi della proteina fibrillare amiloide dovrebbe essere associato alla distruzione degli enzimi che controllano la sintesi delle proteine fibrillari. Si ritiene che il difetto primario nell'amiloidosi sia una violazione della sintesi delle nucleoproteine, che indica la vicinanza dei meccanismi sottili dell'oncologia e dell'amiloidogenesi. La base per la distorsione della funzione cellulare, che porta alla sintesi di proteine fibrillari anomale, è una mutazione del gene responsabile della produzione della proteina.
Amiloidosi genetica, che si sviluppa in connessione con una mutazione genetica, questa amiloidosi è classificata come una malattia poligenica, il cui sviluppo dipende dalla violazione non solo di un enzima, ma del loro complesso.
L'amiloide si forma in quei tumori geneticamente dipendenti: cancro midollare della tiroide, adenomatosi endogene multiple, tumori nelle malattie da immunodeficienza.
I disturbi umorali (disproteinemia, perversioni dell'immunogenesi), così come quelli cellulari (sintesi della proteina amiloide), possono insorgere dopo una particolare malattia o essere la sua espressione (tumore), possono essere una manifestazione di crescenti cambiamenti nel metabolismo della struttura dei tessuti o essere ereditario, che riflette la struttura individuale del metabolismo proteico.
Pertanto, il meccanismo mutazionale porta alla comparsa di amiloidoblasti che secernono una specifica proteina amiloide.
Morfogenesi dell'amiloidosi.
La morfogenesi dell'amiloidosi è costituita dai seguenti collegamenti:
1. Trasformazioni cellulari del RES con comparsa di un clone di cellule capaci di sintetizzare la proteina fibrillare amiloide.
2. Sintesi da parte degli amiloidoblasti del componente principale dell'amiloide: proteina fibrillare (fibrilla).
3. Aggregazione di fibrille con formazione di una "ossatura" della sostanza amiloide.
4. Connessione di fibrille aggregate con proteine plasmatiche e glucoproteine, nonché con MPS acido e formazione di un complesso glucoproteina - amiloide.
Le trasformazioni cellulari del RES costituiscono l'essenza dello stadio pre-amiloide, che è stato studiato nell'amiloidosi secondaria.
Lo stadio pre-amiloide è uno stadio pironinofilo, caratterizzato dalla plasmatizzazione degli organi RES (milza, midollo osseo, linfonodi e fegato) e di altri organi (glomeruli renali, tessuto connettivo peribronchiale, stroma miocardico). Le cellule pironinofile ricche di RNA sono caratterizzate da un'elevata attività degli enzimi redox (glucosio-6-fosfato deidrogenasi). Man mano che la pironinofilia diminuisce, aumenta la quantità di enzimi idrolitici nelle cellule (cellule PAS - positive).
Le trasformazioni cellulari del RES nello stadio pre-amiloide sono dovute a due classi di cellule: plasmociti e linfociti.
Nella milza, insieme alle plasmacellule, che predominano in questa fase, aumenta il numero di cellule reticolari con EPS granulare iperplastico e molti polisomi.
Nel fegato, le plasmacellule e le cellule simili ai linfociti si trovano costantemente nei sinusoidi.
Nel rene, le plasmacellule e le cellule simili ai linfociti sono visibili nello stroma intertubulare.
Pertanto, le trasformazioni cellulari nello stadio pre-amiloide sono dovute all'iperplasia delle cellule plasmatiche e simili ai linfociti.
Sintesi della proteina amiloide fibrillare da parte di cellule di origine mesenchimale.
1. fibroblasti, vicini alla cellula reticolare, responsabili della produzione della proteina reticolina;
2. fibroblasti che producono la proteina del tessuto connettivo - tropocollagene.
Oltre ai fibroblasti, la sintesi delle fibrille amiloidi è associata alle cellule endoteliali.
Le fonti degli amiloidoblasti sono:
1. Nella milza - cellule reticolari e plasmatiche.
2. Nel fegato - cellule di Kupffer che, in determinate condizioni, sono in grado di sintetizzare un'altra proteina fibrillare: il tropocollagene.
3. Nei reni (loro glomeruli), la deposizione di fibrille amiloidi si riscontra più spesso nel mesangio; Le cellule mesangiali diventano amiloidoblasti, che normalmente svolgono il ruolo di fibroblasti modificati che secernono tropocollagene nella membrana basale dei capillari glomerulari, oltre al ruolo di macrofago.
L'aggregazione delle fibrille amiloidi con la formazione di una “struttura” di sostanza amiloide dipende da fattori umorali e tissutali (fattori cellulari).
Tra i fattori umorali sono importanti i legami disolfuro e i gruppi sulfidrilici dell'amiloide, che partecipano all'aggregazione delle proteine in macrostrutture stabili.
Con la formazione di fibrille amiloidi, che è una proteina anomala, le reazioni di riassorbimento cellulare - fibrilloclasia - sono naturali.
Si verifica una sorta di combattimento tra la sintesi della proteina amiloide fibrillare e il suo riassorbimento, un combattimento tra amiloidoblasti e amiloidoclasti. Termina a favore della sintesi della proteina fibrillare amiloide, che si spiega con lo sviluppo della tolleranza alla proteina amiloide. La tolleranza alla proteina della fibrilla amiloide, la cui espressione è un'amiloidoclasia insufficiente e incompetente, può essere considerata una delle ragioni principali dell'accumulo di sostanza amiloide nei tessuti.
Gli amiloidoclasti sono ultrastrutturalmente diversi dagli amiloidoblasti. Il loro citoplasma è privo di RE granulare, ribosomi e mitocondri; il RE liscio appare raramente. Il citoplasma è pieno di inclusioni con una membrana a ciclo singolo contenente fibrille amiloidi. Le inclusioni riflettono le fasi di “digestione” della proteina amiloide fibrillare (le fibrille sono caratterizzate da diverse localizzazioni, fagolisosomi, goccioline lipidiche).
Nella milza, il ruolo degli amiloidoclasti è svolto da macrofagi liberi e fissi di origine ematogena.
Il fegato contiene cellule di Kupffer.
Pertanto, l'aggregazione delle fibrille amiloidi è associata a due processi multidirezionali accoppiati: la sintesi delle fibrille e il loro riassorbimento.
La predominanza dei processi sintetici su quelli di riassorbimento (amiloidoblasti sugli amiloidoclasti) dovuta al ripristino della tolleranza immunologica alla proteina amiloide determina la possibilità della formazione di una struttura fibrillare della sostanza amiloide.
La combinazione delle fibrille amiloidi con le proteine e glicoproteine del plasma e dei tessuti acidi MPS rappresenta lo stadio finale della formazione della sostanza amiloide.
La formazione dell'amiloide avviene all'esterno delle cellule, in stretta connessione con le fibre del tessuto connettivo - reticolare o collagene. Il rapporto tra la sostanza amiloide e le diverse fibre è associato alla partecipazione di diverse cellule (plasmacellule, cellule reticolari o fibroblasti) nella costruzione della proteina amiloide fibrillare.
Esistono due tipi di amiloide a seconda della sua relazione con le strutture fibrillari del tessuto connettivo: perireticolare e pericollagene.
Per l'amiloide perireticolare, che cade lungo le membrane contenenti reticolina di vasi e ghiandole, nonché lungo lo stroma reticolare degli organi parenchimali, danni alla milza, al fegato, ai reni, alle ghiandole surrenali, all'intestino e all'intima dei tessuti piccoli e medi vasi di grandi dimensioni sono tipici.
L'amiloide del pericollagene, che si forma lungo le fibre di collagene, è caratterizzata da un danno predominante all'avventizia dei vasi di grandi e medie dimensioni, allo stroma miocardico, ai muscoli striati e lisci, ai nervi e alla pelle (amiloidosi mesenchimale).
Nella fase finale dell'amiloidogenesi, la permeabilità vascolare aumenta, facilita la connessione delle proteine fibrillari amiloidi e dei tessuti MPS con le proteine e le glucoproteine del plasma sanguigno e determina la presenza di “additivi” ematogeni nell'amiloide.
L'amiloidosi progressiva porta al fallimento funzionale dell'organo, portando all'atrofia dei suoi elementi parenchimali e alla sclerosi. L'organo aumenta di volume, diviene denso e fragile, e al taglio ha un aspetto ceroso o untuoso; alla fine si sviluppano le rughe amiloidi.
L'amiloidosi può essere generalizzata (amiloidosi generale, diffusa) o locale (locale).
Classificazione dell'amiloidosi.
Nel 1929 Lubarsh scriveva che l'amiloidosi primaria differisce dall'amiloidosi secondaria per l'assenza di una malattia “causale” precedente o concomitante, di danni più frequenti ai tessuti mesodermici rispetto a quelli parenchimali (sistema cardiovascolare, tratto digestivo, muscolatura striata e liscia della pelle). . Le cause dell'amiloidosi primaria sono sconosciute, sebbene venga presa in considerazione la somiglianza tra i tipi di amiloidosi ereditaria e primaria.
Tabella 1.
Classificazione dell'amiloidosi Heller et al. (1984).
| Modulo | Tipo di depositi | amiloidosi |
| amiloidosi | perireticolare | pericollagene |
Genetico (ereditario) |
Febbre mediterranea familiare (malattia periodica). Amiloidosi con febbre, orticaria e sordità (forma di Muckle e Wells) | Amiloidosi neuropatica con danno predominante alle estremità inferiori (Andrade, Horta) o superiori (Rukavina). Amiloidosi cardiopatica (Frederiksen). |
Acquisita (secondario) |
Associato a infezioni croniche, malattie del collagene, tumori maligni. | Associato al mieloma multiplo |
| Idiopatico (primario) | Nefropatico |
Classico (sistema). Neuropatico. Cardiopatico. Locale. |
Le cause dell'amiloidosi secondaria possono essere la tubercolosi, che compete con i processi purulento-distruttivi nei polmoni, e anche l'artrite reumatoide e l'osteomielite svolgono un ruolo importante nella comparsa dell'amiloidosi. L'amiloidosi nel mieloma multiplo (plasmocitoma) è isolata come forma indipendente, poiché occupa una posizione intermedia tra secondaria e primaria: poiché nel secondario esiste una malattia "causale" - il mieloma, tuttavia, la natura della distribuzione (vaso, localizzazione) e le proprietà chimico-tintoriali della sostanza amiloide sono più vicine alla forma primaria di amiloidosi.
Alcuni ricercatori ritengono che il meccanismo di formazione dell'amiloide nel mieloma multiplo, a differenza dell'amiloidosi tipica, sia più semplice e si riduca alla diffusione della proteina Bence-Jones sierica micromolecolare nel tessuto, che entra in una "proteina-proteina" o "proteina -polisaccaridi”, che porta alla precipitazione della para-amiloide insolubile.
Tuttavia, attualmente vi sono prove a favore del fatto che nel plasmocitoma la proteina amiloide fibrillare è costituita da catene leggere di immunoglobuline secrete dalla cellula del mieloma, mentre lo stesso meccanismo di formazione si osserva nell'emoblastosi paraproteinemica.
Nel 1961 Briggs completò la classificazione dell’amiloidosi:
1) Amiloidosi primaria (assenza di malattie predisponenti)
a) generalizzato
b) famiglia
c) vie respiratorie (tumorali, nodulari e diffuse)
2) Amiloidosi secondaria (presenza di una malattia predisponente)
3) Amiloidosi senile (cuore)
4) Amiloidosi nel mieloma multiplo
5) Amiloidosi localizzata di tipo tumorale (eccetto l'amiloidosi delle vie respiratorie).
Gafni nel 1964 ridusse le caratteristiche cliniche e morfologiche dell'amiloidosi ereditaria a tre tipi:
1) nefropatico, manifestato dalla sindrome nefrosica con uremia nella fase finale della malattia
2) neuropatico, caratterizzato da polineurite con atrofia muscolare, impotenza, disturbi intestinali e cachessia
3) cardiopatico, caratterizzato da insufficienza cardiaca.
Sulla base di materiali recenti sullo studio dell’amiloidosi,
Serov V.V. propone uno schema di classificazione dell’amiloidosi che tiene conto:
1) La forma dell'amiloidosi a seconda del fattore causale;
2) Possibile meccanismo patogenetico;
3) Tipo clinico di amiloidosi, a seconda della predominanza delle lesioni di un particolare organo o sistema;
4) Aspetto morfogenetico dovuto alle caratteristiche della localizzazione perireticolare o pericollagena della sostanza amiloide;
5) Variante clinica e morfologica dell'amiloidosi - parenchimale o mesenchimale.
Esistono numerose caratteristiche comuni che accomunano l’amiloidosi nel suo insieme. Si tratta, in primo luogo, della disproteinemia, che è espressione di un metabolismo compromesso e possibilmente delle caratteristiche del processo di rinnovamento delle proteine corporee; in secondo luogo, il ruolo uniforme della trasformazione delle cellule RPE nell'emergere della struttura fibrillare dell'amiloide, indipendentemente dal fatto che questa trasformazione sia reattiva, neoplastica o geneticamente determinata; in terzo luogo, cambiamenti submicroscopici negli elementi del sistema del tessuto connettivo che precedono la comparsa dell'amiloide; in quarto luogo, un'unica struttura submicroscopica di amiloide.
Tabella n. 2. Classificazione dell'amiloidosi.
Principali meccanismi di sviluppo |
Tipo clinico |
Aspetto morfologico |
Variante clinica e morfologica |
|
| Idiopatico (primario) | Sconosciuto | Sistema Cardiopatico Neuropatico Nefropatico Enteropatico Epapatico |
Pericollageno Perireticolare |
Mesenchimale Parenchimatoso |
| Ereditario (genetico) | ||||
| 1) malattia periodica (febbre mediterranea familiare) | Nefropatico | >> | >> | |
2) amiloidosi familiare con febbre, orticaria, sordità |
Difetto genetico nella sintesi delle proteine fibrillari del corpo (ereditario fermentopatia) |
>> | >> | >> |
| 3) amiloidosi familiare con manifestazioni allergiche, febbre e nefropatia) | >> | >> | ||
| 4) amiloidosi neuropatica familiare | Neuropatico | Pericollageno | Mesenchimale | |
| tipo I, portoghese | ||||
| 1 | 2 | 3 | 4 | 5 |
| tipo III | ||||
| tipo IV | ||||
| 5) amiloidosi cardiopatica familiare | Cardiopatico | >> | >> | |
Acquisita (secondario) 1) amiloidosi come complicanza di infezioni croniche, malattie del collagene e tumori maligni |
Violazione immunologico omeostasi |
Nefropatico Epinefropatico Epatopatico Misto |
Perireticolare Pericollageno |
Parenchimatoso Mesenchimale |
| 2) paraamiloidosi | Trasformazione neoplastica delle cellule del sistema proteico-sintetico | |||
| Amiloidosi senile | Disturbi involutivi del metabolismo proteico. braditrofia legata all’età |
Cardiopatico | >> | >> |
| Locale simile a un tumore | Sconosciuto |
Un team di ricercatori dell'Università statale di New York a Stony Brook, guidato da William Van Nostrand, Ph.D., professore nel dipartimento di neurochirurgia, ha trovato in un modello di morbo di Alzheimer che in precedenza si accumulavano piccole proteine, note come beta- l'amiloide, nei vasi sanguigni del cervello, può portare a un deterioramento cognitivo precoce, riporta il sito.
I risultati, pubblicati nell'attuale numero online del Journal of Alzheimer's Disease, suggeriscono che l'accumulo di beta-amiloide in stadio iniziale nei vasi sanguigni cerebrali potrebbe essere una potenziale strategia di trattamento per la malattia in stadio iniziale.
La malattia di Alzheimer è una condizione neurodegenerativa che causa un progressivo declino cognitivo. Gli studi hanno dimostrato che nel morbo di Alzheimer e nei disturbi correlati, l'amiloide-beta si accumula nel cervello, il che, secondo i ricercatori, contribuisce alla disfunzione e all'eventuale morte delle cellule nervose. Durante la malattia di Alzheimer, l'amiloide-beta si accumula, formando depositi di amiloide attorno alle cellule nervose note come placche amiloidi o nei vasi sanguigni del cervello.
Nella loro tesi, "La patologia cerebrale microvascolare piuttosto che quella parenchimale della proteina amiloide-β promuove il deterioramento cognitivo precoce nei topi transgenici", il team di Stonybrook ha confrontato due modelli della malattia: uno in cui si sviluppavano placche amiloidi e uno in cui si formavano depositi vascolari di amiloide-beta. vasi del cervello. Il team ha valutato le prestazioni cognitive nel tempo e ha scoperto che nell’arco di un periodo di tre mesi, il modello con beta-amiloide nei vasi sanguigni ha sviluppato un deterioramento cognitivo, mentre il modello con placche amiloidi non ha mostrato alcun deterioramento cognitivo.
"I nostri risultati sono stati piuttosto interessanti perché l'amiloidosi cerebrale, piuttosto che le placche amiloidi attorno alle cellule nervose, ha un impatto precoce sul declino cognitivo", ha affermato il dott. Van Nostrand. "Questa scoperta apre la porta a ulteriori ricerche sul ruolo dell'amiloide nei vasi sanguigni cerebrali nella malattia di Alzheimer e potrebbe essere il primo passo verso lo sviluppo di trattamenti più efficaci per il deterioramento cognitivo basati su questo tipo di amiloide e sulle patologie associate".
È interessante notare che il deterioramento cognitivo in entrambi i modelli ha iniziato a progredire al sesto mese. Ciò suggerisce solo una cosa: quando l’amiloide continua ad accumularsi attorno alle cellule nervose e ai vasi sanguigni nel cervello, si verifica un deterioramento cognitivo.
Il dottor Van Nostrand ha avvertito che per raggiungere una conclusione sarebbero necessari molti più studi che utilizzino diversi modelli comparativi di amiloide. Dopodiché sarà possibile affermare con sicurezza che la deposizione di beta-amiloide nei vasi sanguigni del cervello è un obiettivo chiave nel trattamento della malattia in fase iniziale. Sebbene l'accumulo di amiloide sia una patologia associata al morbo di Alzheimer, dato il complesso processo patologico, l'impatto relativo di ciascuna di queste lesioni amiloidi nel cervello sul deterioramento cognitivo non è ancora del tutto noto.
- In contatto con 0
- Google+ 0
- OK 0
- Facebook 0






