L'emoglobinuria parossistica notturna è una malattia molto rara del gruppo delle anemie emolitiche e non è considerata ereditaria. Si acquisisce durante la vita, sebbene abbia una base genetica. L'essenza della patologia sono i cambiamenti nella struttura delle cellule del sangue (principalmente globuli rossi), che portano alla distruzione prematura delle loro membrane e alla rottura intravascolare (emolisi).
La prevalenza è di circa 16 casi per milione di abitanti e l'incidenza annuale è di 1,3 per milione. Le persone di età compresa tra 20 e 40 anni sono più spesso colpite; non è stata identificata alcuna dipendenza dal genere.
Nel nome sono racchiusi i nomi di ricercatori e medici italiani che hanno dedicato anni allo studio: malattia di Marchiafava-Micheli, malattia di Strübing-Marchiafava.
Cos’è l’“emoglobinuria” e cosa la causa?
L'emoglobinuria è un sintomo di varie malattie che causano la rottura dei globuli rossi per effetto della loro membrana, mentre l'emoglobina lascia le cellule ed entra nel plasma.
In una persona sana, non può superare il 5% del volume totale del plasma sanguigno. Un aumento del livello di emoglobina del 20-25% si osserva nelle malattie congenite o nelle emoglobinopatie (β-talassemia, distruzione dei globuli rossi nell'anemia falciforme).
L'emoglobinuria grave è causata da condizioni in cui i livelli di emoglobina consentiti vengono significativamente superati a causa dell'emolisi dei globuli rossi. Il sistema dei macrofagi non è in grado di elaborare un volume così grande di pigmento e l'emoglobina entra nelle urine.
Le cause dell’emoglobinuria possono essere:
- malattia infettiva acuta (influenza);
- polmonite;
- infortuni;
- intossicazione per avvelenamento con coloranti all'anilina, acido fenico, sale di Berthollet;
- grave ipotermia;
- stress fisico forte e prolungato;
- trasfusione di diversi tipi di sangue;
- ustioni estese;
- È stato stabilito il ruolo della mutazione acquisita del gene PIG-A.
I coloranti all'anilina sono ampiamente utilizzati nell'industria tessile, nella decorazione batik, nei servizi di lavaggio a secco e di tintura, lavorare con loro richiede cautela
Non esiste emoglobinuria senza un elevato livello di emoglobina nel sangue (emoglobinemia). I parossismi prima dell’alba sono associati a uno spostamento fisiologico dell’equilibrio acido-base verso l’acidosi notturna. L'aumento del contenuto di prodotti di degradazione contribuisce ulteriormente all'acidificazione del corpo e all'aumento della degradazione delle cellule del sangue.
Patogenesi dei disturbi
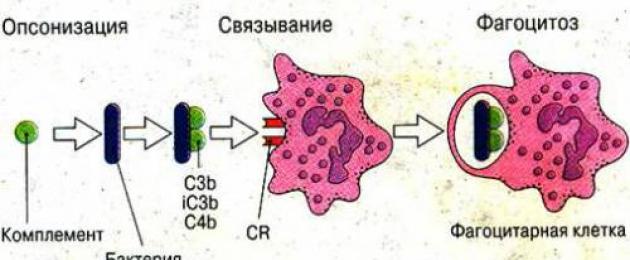
I principali cambiamenti nell'emoglobinuria parossistica notturna si verificano a livello del complemento. Rappresenta una catena di reazioni biochimiche che forniscono l'immunità innata.
Il principio attivo è considerato il complesso di attacco della membrana formato. Contiene circa 30 componenti del regolatore. La sintesi dei componenti del complemento dipende dai segnali ricevuti dai sistemi nervoso ed endocrino. Normalmente è controllato da proteine speciali che non consentono la distruzione delle cellule ospiti (umane).
Con l'emoglobinuria notturna, questo processo viene perso. Lo strato lipidico della membrana cellulare dei globuli rossi viene distrutto, provocandone la morte. È stata dimostrata una maggiore sensibilità della membrana eritrocitaria ai componenti del complemento.

Il complemento è necessario per proteggere le cellule dagli agenti infettivi e utilizzare i prodotti di decomposizione dei microrganismi e delle loro stesse cellule danneggiate.
Anche altre cellule del sangue (leucociti e piastrine) reagiscono provocando difetti nella membrana. Su di essi non è stato riscontrato alcun accumulo di immunoglobuline, il che dimostra l'assenza di un meccanismo di autoallergia e parla a favore di un danno alla cellula precursore comune. È lei che riceve l'informazione genetica (ordine) sull'azione distruttiva.
La regione genetica mancante della cellula staminale è chiamata GPI-AP. La sua carenza nel clone eritrocitario contribuisce alla suscettibilità all'emolisi sotto l'influenza del complemento. Allo stesso tempo, nel corpo può esistere un normale clone di globuli rossi.
L'emoglobinuria parossistica notturna si verifica solo se il clone patologico prevale su quello normale. I globuli rossi di un clone con assenza parziale o completa di GPI-AP vengono rilevati nei pazienti mediante citometria a flusso. È importante che il numero di cellule patologiche nei pazienti non sia lo stesso.
L'aumento della formazione di trombi nella malattia di Marchiafava-Miceli è associato alla stimolazione della coagulazione del sangue da parte di fattori rilasciati durante la distruzione dei globuli rossi.
Forme della malattia
La classificazione delle forme cliniche tiene conto dei dati di laboratorio e del rapporto causa-effetto dei cambiamenti del sangue. È consuetudine distinguere le seguenti varietà:
- Subclinico: non ci sono segni di laboratorio di emolisi; solo metodi altamente sensibili possono rilevare un piccolo numero di cellule prive di GPI-AP. Non esiste un quadro clinico della malattia. Spesso combinato con l'anemia aplastica.
- Classico: sono presenti tutti i sintomi clinici, si manifesta con esacerbazioni periodiche, oltre agli eritrociti, sono colpiti i leucociti e le piastrine, i segni di emolisi vengono determinati in laboratorio (crescita dei reticolociti, enzima sierico lattato deidrogenasi, bilirubina, con un livello ridotto di aptoglobina). Non sono state osservate anomalie dell'ematopoiesi nel midollo osseo.
- Causato dall'insufficienza dell'ematopoiesi del midollo osseo in varie malattie- si presume una patologia concomitante o precedente del midollo osseo con un disturbo dell'ematopoiesi (con anemia aplastica, sindrome mielodisplastica). L'analisi e i risultati clinici rivelano tutte le manifestazioni di emolisi sullo sfondo di anomalie nell'ematopoiesi del midollo osseo.
Secondo un'altra classificazione, si propone di distinguere:
- forma idiopatica o emoglobinuria parossistica notturna stessa;
- patologia sotto forma di sindrome per varie malattie;
- una specie raramente osservata che si verifica dopo l'ipoplasia del midollo osseo.
Nessuna classificazione si basa su un indicatore quantitativo della prevalenza di un clone anomalo nel sangue. È stato dimostrato che è possibile un decorso subclinico con una sostituzione del 90% delle cellule normali. E in altri pazienti, la trombosi grave si verifica in presenza solo del 10% dei globuli rossi alterati.
Sintomi e decorso clinico
La malattia può iniziare improvvisamente (acutamente) o avere un decorso cronico graduale. I periodi di esacerbazione sono chiamati crisi emolitiche. Spesso sono preceduti da un precedente raffreddore, da un'associazione con un'infezione o dal contatto con sostanze tossiche.
I principali sintomi dell’emoglobinuria parossistica notturna includono:
- mal di stomaco;
- dolore al petto di varia intensità e localizzazione - il dolore di diversa localizzazione è associato alla trombosi di piccoli rami del letto arterioso e alla formazione di focolai di ischemia negli organi interni;
- segni di anemia (debolezza, vertigini, mal di testa) - causati da una maggiore distruzione e produzione insufficiente di globuli rossi, inoltre, gli studi indicano una carenza di ferro e acido folico nel sangue dei pazienti;
- giallo della pelle e della sclera - un indicatore del rilascio di bilirubina diretta nel sangue, elaborata dal fegato dall'eccesso di emoglobina;
- disturbo della deglutizione;
- disfunzione erettile negli uomini - si manifesta non solo sullo sfondo delle crisi, ma diventa cronico, causato da una ridotta concentrazione di ossido nitrico nel plasma, alterazione del tono muscolare e vascolare.
- aumento della fatica;
- mancanza di respiro, palpitazioni;
- segni locali di tromboflebite (arrossamento della pelle sopra la vena, gonfiore, dolore alla palpazione, aumento della temperatura);
- Durante l'esame di un paziente, il medico può notare un ingrossamento del fegato e della milza; questo segno è particolarmente importante per diagnosticare lo sviluppo di trombosi e attacchi cardiaci in essi.
Il decorso cronico della malattia contribuisce allo sviluppo di:
- ipertensione polmonare con trombosi nei rami dei vasi polmonari;
- insufficienza renale cronica causata dalla deposizione del prodotto di degradazione dell'emoglobina (emosiderina) nei tubuli renali, trombosi vascolare con formazione di microinfarti;
- elevata sensibilità alle infezioni associate.
Queste sindromi diventano le cause più probabili di morte.
Diagnostica di laboratorio
La diagnosi della malattia di Marchiafava-Micheli viene effettuata dopo un esame approfondito presso centri ematologici che sono in grado di effettuare esami e analisi specifiche.
Nel sangue periferico si trovano:
- eritropenia, leucopenia, trombocitopenia (lo stato di inibizione della crescita generale delle cellule del sangue è chiamato pancitopenia);
- reticolocitosi;
- aumento del livello di emoglobina plasmatica;
- diminuzione dei livelli di ferro e folati.
L’esame del midollo osseo rivela:
- segni di attivazione dell'eritropoiesi (produzione di globuli rossi) dovuta all'accumulo di cellule precursori (normoblasti, plasma e mastociti);
- diminuzione del numero di granulociti e megacariociti;
- aree di emorragia, accumulo di globuli rossi emolizzati nei seni;
- nella fase di soppressione dell'ematopoiesi sono visibili zone di degenerazione grassa e devastazione.
Test specifici basati sulla maggiore sensibilità degli eritrociti difettosi al complemento nelle condizioni più favorevoli in termini di composizione del mezzo sono i test di Hem (acido) e di Hartmann (con saccarosio).
Entrambi i test verificano la "sopravvivenza" dei globuli rossi in un campione di sangue posto in una soluzione debole. Il test di Hem è positivo quando la distruzione è pari o superiore al 5%, mentre quello di Hartman è pari o superiore al 4%.
Il test di Coombs viene eseguito per escludere una connessione con il meccanismo autoimmune di distruzione cellulare; è negativo per l'emoglobinuria notturna.

La colorazione dell'urina indica un contenuto significativo di ossiemoglobina in essa contenuta.
Un esame delle urine ha mostrato che uno dei primi segni di emoglobinuria notturna sono le urine del mattino e della notte, di colore rosso scuro. Nel tempo, l'urina raccolta si separa in strati:
- il liquido sopra è trasparente, ma conserva il colore;
- le particelle di cellule morte di origine organica vengono determinate dal basso.
Da quali malattie bisogna distinguere l’emoglobinuria notturna?
La diagnosi differenziale dell'emoglobinuria parossistica notturna viene effettuata con altre anemie simili nel decorso clinico, principalmente con l'anemia emolitica di tipo autoimmune e con quella aplastica.
Le caratteristiche comuni sono:
- una forte diminuzione del numero di globuli rossi;
- reticolocitosi;
- presenza di ittero;
- febbre;
- aumento della concentrazione di bilirubina libera;
- tendenza alla trombosi;
- moderato ingrossamento del fegato e della milza.
Nell'anemia non si riscontrano livelli elevati di emoglobina nel plasma sanguigno o di urobilina nelle urine. I test di laboratorio di Hem e Hartman sono negativi, ma il test di Coombs è positivo.
La diagnosi è significativamente difficile se la malattia si manifesta sotto forma di crisi temporanee sullo sfondo di una forma acuta di leucemia mieloblastica, eritromielosi, osteomielosclerosi, lesioni metastatiche del midollo osseo nei tumori maligni.

La massa dei globuli rossi viene conservata fredda in confezioni speciali
Trattamento
Ad oggi non esiste un modo efficace per fermare la degradazione dei globuli rossi. Non resta che utilizzare l'opzione sostitutiva e trasfondere il paziente con globuli rossi lavati provenienti da donatori.
Una caratteristica importante è il buon “atteggiamento” del corpo del paziente nei confronti delle cellule estranee iniettate; praticamente non vi è alcuna reazione di rigetto. Considerando la presenza di cellule GPI-AP sane nelle membrane e l’assenza di mutazioni genetiche in esse, è possibile supportare l’ematopoiesi del paziente.
Il sangue utilizzato per la trasfusione deve essere mantenuto congelato per almeno una settimana per distruggere completamente i leucociti in esso contenuti. Una volta raggiunti il paziente, possono causare un'esacerbazione dell'emolisi a causa dell'aumentata sensibilizzazione e attivazione del complemento.
Con trasfusioni frequenti è ancora possibile la formazione di anticorpi antieritrociti. In tali pazienti, la successiva trasfusione viene eseguita dopo diverse procedure di lavaggio dei globuli rossi con soluzione salina e controllo del sangue del donatore mediante il test di Coombs.
Il numero di trasfusioni viene solitamente prescritto almeno cinque, ma dipende dalla gravità delle condizioni del paziente e dalla risposta al trattamento.
Per stimolare la corretta emopoiesi, Nerobol (un farmaco ormonale anabolizzante) viene utilizzato in cicli fino a tre mesi. In questo caso è possibile un cambiamento nello stato funzionale del fegato.
Ai fini del trattamento e della prevenzione della formazione di trombi, viene utilizzata l'eparina, seguita da un passaggio a dosi di mantenimento di anticoagulanti indiretti.
Per compensare le perdite di ferro vengono prescritte compresse.
Un'indicazione per la rimozione della milza può essere un forte aumento delle dimensioni e segni di infarto. La splenectomia viene eseguita raramente.
Per proteggere il fegato vengono prescritti farmaci epatoprotettivi. A volte la terapia steroidea aiuta.

Il farmaco viene somministrato solo per via endovenosa
Negli ultimi anni sono apparse informazioni sull'uso del farmaco Eculizumab (Soliris), ottenuto da anticorpi monoclonali. Secondo i rapporti disponibili, blocca l'emolisi ed è in grado di resistere al complemento sanguigno. Il farmaco è considerato la medicina più costosa al mondo. La sua azione e gli effetti negativi non sono stati sufficientemente studiati.
L'emoglobinuria notturna non ha ancora un trattamento specifico. Anche con una terapia di supporto sufficiente, i pazienti vivono circa cinque anni dopo l’esordio della malattia. Non c'è prevenzione. Tutti dovrebbero attenersi ad un comportamento corretto durante il lavoro e quando si ha un contatto forzato con composti tossici.
L’emoglobinuria parossistica notturna, conosciuta anche come malattia di Strübing-Marchiafava, malattia di Marchiafava-Micheli, è una malattia rara, una patologia ematica progressiva che mette a rischio la vita del paziente. È uno dei tipi di anemia emolitica acquisita causata da disturbi nella struttura delle membrane degli eritrociti. Le cellule difettose sono soggette a decadimento prematuro (emolisi) che avviene all'interno dei vasi sanguigni. La malattia è di natura genetica, ma non è considerata ereditaria.
L’incidenza è di 2 casi ogni milione di persone. L’incidenza è di 1,3 casi per milione di persone all’anno. Si manifesta prevalentemente nelle persone di età compresa tra 25 e 45 anni; non è stata identificata alcuna dipendenza dell'incidenza dal sesso e dalla razza. Ci sono casi isolati della malattia nei bambini e negli adolescenti.
Importante: l'età media alla quale viene diagnosticata la malattia è di 35 anni.
Cause della malattia
Le cause e i fattori di rischio per lo sviluppo della malattia non sono noti. È stato accertato che la patologia è causata da una mutazione nel gene PIG-A, situato nel braccio corto del cromosoma X. Il fattore mutageno non è stato ancora identificato. Nel 30% dei casi di emoglobinuria parossistica notturna esiste una connessione con un'altra malattia del sangue: l'anemia aplastica.
La formazione, lo sviluppo e la maturazione delle cellule del sangue (ematopoiesi) avviene nel midollo osseo rosso. Tutte le cellule del sangue specializzate sono formate dalle cosiddette cellule staminali, non specializzate, che hanno mantenuto la capacità di dividersi. Formate come risultato di successive divisioni e trasformazioni, le cellule del sangue mature entrano nel flusso sanguigno.
Una mutazione nel gene PIG-A anche in una singola cellula porta allo sviluppo della EPN. Il danno al gene modifica anche l'attività delle cellule nei processi di mantenimento del volume del midollo osseo; le cellule mutanti si moltiplicano più attivamente di quelle normali. Nel tessuto emopoietico si forma rapidamente una popolazione di cellule che producono globuli difettosi. In questo caso il clone mutante non è un tumore maligno e può scomparire spontaneamente. La sostituzione più attiva delle cellule normali del midollo osseo con quelle mutanti avviene nei processi di ripristino del tessuto del midollo osseo dopo un danno significativo causato, in particolare, dall'anemia aplastica.
Il danno al gene PIG-A porta a interruzioni nella sintesi delle proteine di segnalazione che proteggono le cellule del corpo dagli effetti del sistema del complemento. Il sistema del complemento è costituito da proteine specifiche del plasma sanguigno che forniscono una protezione immunitaria generale. Queste proteine si legano ai globuli rossi danneggiati, li sciolgono e l'emoglobina rilasciata si mescola con il plasma sanguigno.
Classificazione
Sulla base dei dati disponibili sulle cause e le caratteristiche dei cambiamenti patologici, si distinguono diverse forme di emoglobinuria parossistica notturna:
- Subclinico.
- Classico.
- Associato a disturbi dell'ematopoiesi.
La forma subclinica della malattia è spesso preceduta da anemia aplastica. Non ci sono manifestazioni cliniche della patologia, ma la presenza di un piccolo numero di cellule del sangue difettose viene rilevata solo durante gli esami di laboratorio.
In una nota. Si ritiene che la EPN sia una malattia più complessa, il cui primo stadio è l'anemia aplastica.
La forma classica si manifesta con sintomi tipici; nel sangue del paziente sono presenti popolazioni di globuli rossi, piastrine e alcuni tipi di leucociti difettosi. I metodi di ricerca di laboratorio confermano la distruzione intravascolare di cellule patologicamente alterate; i disturbi dell'ematopoiesi non vengono rilevati.
Dopo aver sofferto di malattie che portano all'insufficienza emopoietica, si sviluppa una terza forma di patologia. Un quadro clinico pronunciato e la lisi intravascolare dei globuli rossi si sviluppano sullo sfondo delle lesioni del midollo osseo.
Esiste una classificazione alternativa, secondo la quale si distinguono:
- In realtà EPN, idiopatico.
- Svilupparsi come sindrome concomitante con altre patologie.
- Svilupparsi come conseguenza dell'ipoplasia del midollo osseo.
La gravità della malattia nei diversi casi non è sempre correlata al numero di globuli rossi difettosi. Sono stati descritti sia casi subclinici con un contenuto di cellule modificate vicino al 90%, sia casi estremamente gravi con sostituzione del 10% della popolazione normale.
Sviluppo della malattia
È attualmente noto che nel sangue di pazienti affetti da emoglobinuria parossistica notturna possono essere presenti tre tipi di eritrociti con diversa sensibilità alla distruzione da parte del sistema del complemento. Oltre alle cellule normali, nel flusso sanguigno circolano globuli rossi, la cui sensibilità è molte volte superiore al normale. Nel sangue dei pazienti con diagnosi di malattia di Marchiafava-Micheli sono state trovate cellule la cui sensibilità al complemento era 3-5 e 15-25 volte superiore al normale.
I cambiamenti patologici colpiscono anche altre cellule del sangue, vale a dire piastrine e granulociti. Al culmine della malattia, i pazienti sperimentano pancitopenia, un numero insufficiente di cellule del sangue di diversi tipi.
La gravità della malattia dipende dal rapporto tra le popolazioni di cellule del sangue sane e difettose. Il contenuto massimo di globuli rossi ipersensibili all'emolisi dipendente dal complemento viene raggiunto entro 2-3 anni dal momento della mutazione. In questo momento compaiono i primi sintomi tipici della malattia.
La patologia di solito si sviluppa gradualmente; l'insorgenza di crisi acute è rara. Le esacerbazioni si verificano sullo sfondo delle mestruazioni, dello stress grave, delle malattie virali acute, della chirurgia, del trattamento con alcuni farmaci (in particolare quelli contenenti ferro). A volte la malattia peggiora quando si mangiano determinati alimenti o senza una ragione evidente.
Esistono prove di manifestazioni della malattia di Marchiafava-Micheli dovute all'esposizione alle radiazioni.
La dissoluzione delle cellule del sangue a vari livelli nei pazienti con emoglobinuria parossistica notturna accertata si verifica costantemente. Periodi di moderata progressione sono intervallati da crisi emolitiche, massiccia distruzione di globuli rossi, che porta ad un netto deterioramento delle condizioni del paziente.
Al di fuori di una crisi, i pazienti sono preoccupati per le manifestazioni di ipossia generale moderata, come mancanza di respiro, attacchi di aritmia, debolezza generale e peggioramento della tolleranza all'esercizio. Durante una crisi compare dolore addominale, localizzato principalmente nella zona dell'ombelico e nella parte bassa della schiena. L'urina diventa nera, la parte più scura è al mattino. Le ragioni di questo fenomeno non sono state ancora stabilite in modo definitivo. Con la EPN si sviluppa una leggera pasosità del viso e si nota il giallo della pelle e della sclera.
La malattia di Marchiafava-Michele, emoglobinuria parossistica notturna con emosiderinuria costante, la malattia di Strübing-Marchiafava è una sorta di anemia emolitica acquisita che si manifesta con emolisi intravascolare costante, emosiderinuria, inibizione della granulo- e trombocitopoiesi.Una nota! Un sintomo tipico della malattia è l'urina macchiata. In circa la metà dei casi conosciuti la malattia non si manifesta.
Nei periodi tra le crisi, i pazienti possono sperimentare:
- anemia;
- tendenza alla trombosi;
- ingrossamento del fegato;
- manifestazioni di distrofia miocardica;
- tendenza all'infiammazione di origine infettiva.
Quando le cellule del sangue vengono distrutte, vengono rilasciate sostanze che aumentano la coagulazione, causando la trombosi. Nei vasi del fegato e dei reni possono formarsi coaguli di sangue; anche i vasi coronarici e cerebrali possono subire danni che possono portare alla morte. La trombosi localizzata nei vasi del fegato porta ad un aumento delle dimensioni dell'organo. I disturbi del flusso sanguigno intraepatico portano a cambiamenti degenerativi nei tessuti. Quando il sistema della vena porta o le vene spleniche sono bloccati, si sviluppa la splenomegalia. I disturbi del metabolismo dell'azoto sono accompagnati da disfunzione della muscolatura liscia; alcuni pazienti lamentano difficoltà di deglutizione, spasmi dell'esofago e negli uomini è possibile la disfunzione erettile.
Importante! Le complicanze trombotiche della EPN colpiscono prevalentemente le vene; raramente si sviluppa trombosi arteriosa.
Video - Emoglobinuria parossistica notturna
Meccanismi di sviluppo delle complicanze della EPN
La crisi emolitica si manifesta con i seguenti sintomi:
- dolore addominale acuto causato da trombosi multipla delle piccole vene mesenteriche;
- aumento dell'ittero;
- dolore nella regione lombare;
- abbassare la pressione sanguigna;
- aumento della temperatura corporea;
- colorazione delle urine nera o marrone scuro.
In rari casi si sviluppa un “rene emolitico”, una specifica forma transitoria di insufficienza renale accompagnata da anuria acuta. A causa della ridotta funzione escretoria, nel sangue si accumulano composti organici contenenti azoto, che sono i prodotti finali della degradazione proteica, e si sviluppa azotemia. Dopo che il paziente si riprende dalla crisi, il contenuto degli elementi formati nel sangue viene gradualmente ripristinato, l'ittero e le manifestazioni di anemia svaniscono parzialmente.
Il decorso più comune della malattia è la crisi, intervallata da periodi di condizioni stabili e soddisfacenti. In alcuni pazienti, i periodi tra le crisi sono molto brevi, insufficienti a ripristinare la composizione del sangue. Tali pazienti sviluppano un'anemia persistente. Esiste anche una variante del decorso con esordio acuto e crisi frequenti. Nel tempo, le crisi diventano meno frequenti. In casi particolarmente gravi, è possibile la morte, causata da insufficienza renale acuta o trombosi dei vasi sanguigni che riforniscono il cuore o il cervello.
Importante! Non sono stati identificati schemi quotidiani nello sviluppo delle crisi emolitiche.
In rari casi la malattia può avere un decorso tranquillo a lungo termine; sono stati descritti casi isolati di guarigione.
Diagnostica
Nelle fasi iniziali della malattia, la diagnosi è difficile a causa della manifestazione di sintomi sparsi e aspecifici. La diagnosi a volte richiede diversi mesi di osservazione. Il sintomo classico, la colorazione specifica delle urine, compare durante le crisi e non in tutti i pazienti. I motivi per sospettare la malattia di Marchiafava-Miceli sono:
- carenza di ferro di eziologia sconosciuta;
- trombosi, mal di testa, attacchi di dolore alla parte bassa della schiena e all'addome senza motivo apparente;
- anemia emolitica di origine sconosciuta;
- fusione delle cellule del sangue, accompagnata da pancitopenia;
- complicanze emolitiche associate alla trasfusione di sangue fresco di donatore.
Nel processo diagnostico è importante stabilire il fatto della rottura intravascolare cronica dei globuli rossi e identificare i segni sierologici specifici della EPN.
In un complesso di studi, se si sospetta emoglobinuria parossistica notturna, oltre agli esami generali delle urine e del sangue, vengono eseguiti quanto segue:
- determinazione del contenuto di emoglobina e aptoglobina nel sangue;
- immunofenotipizzazione mediante citometria a flusso per identificare popolazioni cellulari difettose;
- test sierologici, in particolare il test di Coombs.
È necessaria la diagnosi differenziale con l'emoglobinuria e l'anemia di altre eziologie; in particolare, deve essere esclusa l'anemia emolitica autoimmune. I sintomi più comuni sono anemia, ittero e aumento della bilirubina nel sangue. Non in tutti i pazienti si osserva un ingrossamento del fegato e/o della milza
Segni Emolitico autoimmune
anemiaPNG Prova di Coombs + - Aumento dei contenuti gratuiti
emoglobina nel plasma sanguigno- + Test di Hartmann (saccarosio) - + Test di Hem (acido) - + Emosiderina nelle urine - + Trombosi ± + Epatomegalia ± ± Splenomegalia ± ± I risultati del test di Hartmann e Hem sono specifici per la EPN e rappresentano i segni diagnostici più importanti.
Trattamento
Il sollievo di una crisi emolitica viene effettuato mediante ripetute trasfusioni di globuli rossi, scongelati o precedentemente lavati più volte. Si ritiene che siano necessarie almeno 5 trasfusioni per ottenere un risultato duraturo, tuttavia il numero di trasfusioni può differire dalla media ed è determinato dalla gravità delle condizioni del paziente.
Attenzione! Il sangue non può essere trasfuso a tali pazienti senza previa preparazione. La trasfusione di sangue da donatori aggrava la crisi.
Per l'eliminazione sintomatica dell'emolisi, ai pazienti può essere prescritto Nerobol, ma sono possibili ricadute dopo la sospensione del farmaco.
Inoltre vengono prescritti acido folico, ferro ed epatoprotettori. Quando si sviluppa una trombosi, vengono utilizzati anticoagulanti ad azione diretta ed eparina.
In casi estremamente rari, al paziente viene indicata la splenectomia - rimozione della milza.
Tutte queste misure sono di supporto; alleviano le condizioni del paziente, ma non eliminano la popolazione di cellule mutanti.
0

Cause:
Le cause della malattia sono associate alla distruzione intravascolare dei globuli rossi, che sono in gran parte difettosi. Insieme alla popolazione patologica dei globuli rossi, vengono preservate anche alcune cellule normali che hanno una durata di vita normale. Sono stati rilevati disturbi nella struttura dei granulociti e delle piastrine. La malattia non è ereditaria, ma qualsiasi fattore esterno provoca la formazione di una popolazione cellulare difettosa, che è un clone, cioè la discendenza di una cellula inizialmente modificata non è nota.Le complicanze trombotiche della EPN sono associate all'emolisi intravascolare, che provoca la formazione di trombi. L'origine di un segno importante, ma tutt'altro che obbligatorio della malattia - parossismi di emoglobinuria durante la notte o al mattino - rimane poco chiara.
Il parossismo non è legato all'ora del giorno, ma al sonno, che durante il giorno può provocare anche una crisi. Nella EPN si osserva un'aumentata sensibilità del complemento degli eritrociti patologici. Forse questa è la base per provocare una crisi emolitica con una trasfusione di sangue fresco, che contiene fattori che attivano il complemento. La trasfusione di sangue conservato per più di una settimana non provoca emolisi.
Sintomi dell'emoglobinuria parossistica notturna:
La malattia si sviluppa lentamente: compaiono segni di moderata anemia, debolezza, affaticamento, palpitazioni durante l'esercizio e dolore addominale, spesso associati a trombosi dei vasi mesenterici. La pelle e le mucose sono itteriche pallide, grigiastre a causa dell'anemia e dei depositi di emosiderina. Segni caratteristici dell'emolisi intravascolare.La comparsa di urina nera non è un segno permanente. Poiché la EPN è spesso accompagnata da leucopenia (dovuta principalmente alla granulocitopenia), sono possibili complicanze infettive croniche. La trombocitopenia può essere complicata dalla sindrome emorragica. L'escrezione a lungo termine di emoglobina ed emosiderina nelle urine porta gradualmente allo sviluppo di uno stato di carenza di ferro: si verifica la sindrome astenica, compaiono pelle secca e unghie fragili.
Il quadro ematico è caratterizzato inizialmente da anemia normocromica e poi ipocromica, lieve reticolocitosi (2-4% o più), leucopenia e trombocitopenia. La morfologia degli eritrociti non ha caratteristiche caratteristiche. Nel midollo osseo si osserva iperplasia del germe rosso, ma nel trefino si osserva un leggero aumento della cellularità del midollo osseo, che può diventare ipoplastico con il progredire della malattia.
A causa dell'emolisi intravascolare costantemente in corso, il contenuto di emoglobina libera nel plasma aumenta (normalmente inferiore a 0,05 g/l). I livelli di ferro nel siero sono inizialmente normali ma possono poi essere significativamente ridotti. Insieme all'esordio tipico della malattia, quando prevale la sindrome emolitica, può svilupparsi un quadro di sindrome aplastica, che dopo alcuni anni può essere complicata da una crisi emolitica con tipica emoglobinuria notturna. Più spesso, una crisi emolitica provoca una trasfusione di sangue.
Diagnosi:
La diagnosi viene posta sulla base dei segni di emolisi intravascolare (anemia, lieve reticolocitosi, emosiderina nelle urine). La diagnosi viene chiarita da studi speciali (test del saccarosio positivo, test di Hem, test di Coombs negativo).La forma emolisina dell'anemia emolitica autoimmune, simile nelle manifestazioni esterne alla EPN, si verifica con emolisi intravascolare ed è caratterizzata dalla presenza di emolisina nel siero del sangue e da un test di Coombs positivo. A differenza della EPN, non si verifica leucopenia o trombocitopenia; il prednisolone di solito ha un buon effetto. L'EPN può essere distinta dall'anemia aplastica dal quadro del midollo osseo: con aplasia, il trepanato è caratterizzato da una predominanza di grasso, con emolisi - da iperplasia cellulare, tuttavia, in rari casi di EPN, un quadro di ipoplasia del midollo osseo può si sviluppano, sebbene l'emosiderina sia costantemente rilevata nelle urine e la reticolocitosi nel sangue.
Trattamento dell'emoglobinuria parossistica notturna:
Il trattamento in assenza di anemia grave non viene effettuato. La sindrome anemica grave richiede una trasfusione di globuli rossi; I migliori risultati si ottengono con la trasfusione di eritrociti lavati o invecchiati per 7-10 giorni. Per l'ipoplasia dell'ematopoiesi sono indicati gli steroidi anabolizzanti: Nerobol - 10-20 mg al giorno o Retabolil - 50 mg per via intramuscolare per 2-3 settimane.Vengono utilizzati integratori di ferro, ma a volte possono provocare una crisi emolitica. Per prevenire una crisi, il ferro viene prescritto a piccole dosi durante il trattamento con steroidi anabolizzanti. Per la trombosi è indicata l'eparina: alla prima iniezione si somministrano 10.000 unità per via endovenosa, poi 5-10mila unità 2-3 volte al giorno sotto la pelle dell'addome (con un ago sottile ad una profondità di 2 cm nel grasso tessuto) sotto il controllo della coagulazione del sangue. Controindicazioni al trattamento con eparina sono una recente esacerbazione di un'ulcera gastrica o duodenale, nonché la presenza di fonti di sanguinamento.
L'emoglobinuria parossistica notturna (EPN) è una malattia acquisita che si manifesta con anemia emolitica persistente, emoglobinuria parossistica o persistente ed emolisi intravascolare. La rarità di questo tipo di anemia emolitica è caratterizzata dal fatto che la EPN colpisce 1 persona su mezzo milione, per lo più giovani.
Le cause della malattia sono attualmente sconosciute. Si presume che si verifichi a causa della presenza di un clone anormale di globuli rossi inclini all'emolisi intravascolare. A sua volta, l'inferiorità dei globuli rossi è una conseguenza di difetti strutturali e biochimici nella loro membrana. È noto che in una membrana difettosa viene attivata la perossidazione lipidica, che favorisce la rapida lisi dei globuli rossi; inoltre, nel processo patologico sono coinvolti cloni anormali di granulociti e piastrine. Il ruolo principale nell'insorgenza delle complicanze trombotiche della EPN appartiene alla distruzione intravascolare degli eritrociti e all'avvio della coagulazione del sangue da parte di fattori rilasciati durante questo processo. La EPN, di regola, inizia gradualmente e procede cronicamente con crisi periodiche. Le crisi sono provocate da infezioni virali, interventi chirurgici, stress psico-emotivo, mestruazioni e dall'uso di numerosi farmaci e alimenti.
Sintomi dell'emoglobinuria parossistica notturna
Sintomi della EPN durante una crisi:
- dolore parossistico nella cavità addominale;
- dolore nella regione lombare;
- ittero della pelle e della sclera; ipertermia; pastosità facciale;
- colore nero delle urine, soprattutto di notte;
- un forte calo della pressione sanguigna;
- ingrossamento transitorio della milza;
- cessazione della produzione di urina.
In alcuni casi, la crisi emolitica termina con la morte.
Sintomi della EPN al di fuori della crisi:
- debolezza generale;
- colore della pelle pallido con una tinta itterica;
- anemia;
- tendenza alla trombosi; ematuria; ipertensione; ingrossamento del fegato; dispnea; battito cardiaco; frequenti malattie infettive.
Diagnostica
- Esame del sangue: anemia (normocromica, successivamente ipocromica), leucocitopenia e trombocitopenia moderate, il livello di ferro nel siero è significativamente ridotto.
- Esame delle urine: colorazione nera, emoglobinuria, emosiderinuria, proteinuria. Il test della benzidina nelle urine di Gregersen è positivo.
- Il test specifico di Ham è positivo.
- Il test specifico di Hartmann è positivo.
- Puntura del midollo osseo: iperplasia della linea emopoietica rossa, ma nei casi più gravi si può osservare anche ipoplasia del midollo osseo e aumento della quantità di tessuto adiposo nel midollo osseo.
Trattamento dell'emoglobinuria parossistica notturna
Il trattamento della EPN è sintomatico e consiste principalmente in trasfusioni di sangue sostitutive, la cui quantità e frequenza dipendono dalla “risposta” a queste misure. Nel trattamento della EPN, il metandrostenolone viene utilizzato alla dose di 30-50 mg/die per almeno 2-3 mesi. La lotta contro l'ipoplasia del midollo osseo viene effettuata mediante l'uso endovenoso di immunoglobulina antitimocitaria alla dose di 150 mg/die per 4-10 giorni. Si consiglia di assumere integratori di ferro per os in piccole dosi. A volte i corticosteroidi ad alti dosaggi hanno un buon effetto. L'ipoplasia del midollo osseo con lo sviluppo di complicanze trombotiche sono indicazioni per il suo trapianto. Sono stati descritti casi isolati di guarigione dalla EPN; in alcuni casi, la durata di un decorso favorevole della malattia è di diversi decenni.
Farmaci essenziali
Ci sono controindicazioni. È necessaria una consulenza specialistica.
L'anemia aplastica è una malattia rara del sistema sanguigno, caratterizzata da pancitopenia nel sangue periferico e nel midollo osseo ipocellulare (fino all'aplasia completa) con sostituzione del tessuto emopoietico attivo con tessuto adiposo. La prima descrizione della malattia, fatta da P. Ehrlich, risale al 1888.
La malattia si manifesta nella maggior parte delle regioni d'Europa e d'America con una frequenza di 2-3 casi all'anno per 1 milione di abitanti. L’incidenza dell’anemia aplastica è 2-3 volte maggiore nell’Asia orientale. Si registrano due picchi di incidenza: tra i 10 ei 25 anni e nelle persone con più di 60 anni, senza differenze significative per genere. Una forma rara è l'anemia aplastica congenita - anemia di Fanconi, che nella maggior parte dei casi si manifesta come malattia autosomica recessiva.
Eziologia e patogenesi
L'eziologia della malattia nel 70-80% dei casi è sconosciuta (forme idiopatiche), mentre in altri casi l'insorgenza dell'anemia aplastica è associata a diversi fattori chimici, fisici, infezioni (anemia aplastica post-epatite, forme associate a citomegalovirus, infezione da parvovirus, ecc.).
Le più comuni sono le forme acquisite di anemia aplastica, ma fino al 15-20% dei casi della malattia possono essere varianti costituzionali/congenite (anemia di Fanconi, anemia associata a discheratosi), accompagnate da diverse anomalie citogenetiche. Esiste anche una variante dell'anemia aplastica associata ad emoglobinuria parossistica notturna.
Il principale meccanismo patogenetico per lo sviluppo dell'aplasia ematopoietica nell'anemia aplastica è il danno immunomediato alle cellule staminali ematopoietiche. Allo stesso tempo, non si può escludere un difetto funzionale delle cellule staminali ematopoietiche e una patologia del microambiente ematopoietico.
La prova di processi immunitari attivi nel midollo osseo dei pazienti con anemia aplastica è un aumento del contenuto di linfociti T maturi e attivati, cellule con un fenotipo soppressore-killer e un'inversione del rapporto helper-soppressore, che vengono rilevati naturalmente in questo gruppo di pazienti.
Caratterizzato da un aumento del livello di citochine che influenzano negativamente i processi ematopoietici, come IFN, IL-2, fattore di necrosi tumorale (TNFα). Allo stesso tempo, anche il meccanismo di attivazione incontrollato potenziato dell’apoptosi Fas-dipendente delle cellule ematopoietiche sembra svolgere un ruolo significativo nello sviluppo della malattia. I pazienti affetti da anemia aplastica non sono solitamente caratterizzati da un deficit di fattori che regolano l'ematopoiesi. Esistono alcuni collegamenti patogenetici tra anemia aplastica, emoglobinuria parossistica notturna e sindrome mielodisplastica, la cui natura non è ancora del tutto chiara. L'anemia aplastica può eventualmente trasformarsi in emoglobinuria parossistica notturna e sindrome mielodisplastica. Un piccolo clone EPN senza segni di emolisi viene rilevato, secondo studi recenti, nel 50-70% dei pazienti con anemia aplastica. Cloni con anomalie citogenetiche, in assenza di prove a favore della sindrome mielodisplastica, possono essere rilevati in alcuni pazienti con anemia aplastica.
Quadro clinico
In termini di gravità del decorso, elevata mortalità precoce tra i pazienti con grave anemia aplastica e complessità del trattamento, questa categoria è paragonabile al gruppo di pazienti con leucemia acuta. La mortalità senza trattamento nei primi 6 mesi nelle forme gravi di anemia aplastica raggiunge il 50% o più. Le cause della morte dei pazienti sono la progressione della malattia e lo sviluppo di complicanze emorragiche e gravi infettive.
Le manifestazioni cliniche della malattia sono causate principalmente dalla presenza di sindrome anemica ed emorragica. I pazienti con anemia aplastica sono caratterizzati da pallore della pelle e mucose visibili a vari gradi di gravità. Di norma, si verificano emorragie di varie dimensioni sulla pelle e sulle mucose, da puntiformi a confluenti. Spesso si verificano emorragie nel fondo e nella retina dell'occhio, accompagnate da una diminuzione dell'acuità visiva. Le emorragie sulle mucose del cavo orale possono essere accompagnate da sintomi di stomatite e necrotizzazione dei tessuti molli. Nelle forme gravi della malattia con gravi manifestazioni emorragiche, sono possibili emorragie nella parete intestinale. In quest'ultimo caso si verificherà il quadro clinico corrispondente: dolore, gonfiore e dolore alla palpazione, disturbi della peristalsi. Allo stesso tempo, in alcuni pazienti (in media fino al 20%), durante l'esame iniziale non si notano manifestazioni emorragiche visibili.I cambiamenti nel sistema cardiovascolare si manifestano con tachicardia, espansione dei confini del cuore, suoni cardiaci ovattati, e soffio sistolico sopra la superficie del cuore.
Linfoadenopatia, epato- e splenomegalia non sono tipiche dell'anemia aplastica. Con la granulocitopenia profonda, vi è una maggiore tendenza a sviluppare complicanze infettive e infiammatorie-necrotiche.
L'esordio acuto dell'anemia aplastica si osserva nel 12-15% dei pazienti ed è accompagnato da febbre, mal di gola necrotizzante, grave sanguinamento nasale, gengivale, uterino e comparsa di emorragie multiple sulla pelle e sulle mucose. In oltre l'80% dei pazienti, la malattia si sviluppa gradualmente con un aumento delle manifestazioni della sindrome anemica ed emorragica.
Nell'anemia di Fanconi, che di solito viene diagnosticata in giovane età, possono essere rilevate anomalie scheletriche e pigmentazione della pelle - macchie café au lait.
Ricerca di laboratorio
Un emocromo completo solitamente mostra pancitopenia con relativa conservazione dei linfociti. L'anemia è solitamente normocromica e caratterizzata da reticolocitopenia. Si può osservare macrocitosi. Le piastrine sono significativamente ridotte in numero e solitamente di piccole dimensioni.
Il quadro del midollo osseo dei pazienti con anemia aplastica è caratterizzato da un numero ridotto di cellule ematopoietiche e da spazi adiposi allargati. L'eritropoiesi è ridotta o assente; spesso si osserva diseritropoiesi, non accompagnata da alterazioni displastiche in altre emopoiesi, come nella sindrome mielodisplastica. Il numero di megacariociti e cellule granulocitiche è significativamente ridotto. Poiché il danno al midollo osseo non è uniforme, si può osservare un'iperplasia focale delle linee eritroidi e granulocitiche e, dopo l'aspirazione della loro "tasca calda" con un focus di emopoiesi intatta, gli indicatori del mielogramma, specialmente nelle fasi iniziali della malattia, possono essere vicini alla normalità. Per valutare la cellularità complessiva e valutare la morfologia delle cellule ematopoietiche residue, lo studio di un campione bioptico di midollo osseo di alta qualità è cruciale.
Diagnosi differenziale
La diagnosi di anemia aplastica viene effettuata sulla base della determinazione della pancitopenia nel sangue periferico e della ridotta cellularità del midollo osseo mediante trepanobiopsia. Caratteristica è la sostituzione del tessuto emopoietico attivo con tessuto adiposo, in assenza di infiltrazione di cellule atipiche e segni di fibrosi. Un attento esame degli strisci di sangue e dei preparati di midollo osseo consente di escludere la presenza di neutrofili displastici, piastrine anormali e cellule tumorali.
Quando si formula una diagnosi di anemia aplastica, gruppi di ricerca internazionali hanno raccomandato di tenere conto della presenza di almeno due dei seguenti parametri ematici in combinazione con cambiamenti caratteristici del quadro del midollo osseo: livello di emoglobina
Il piano di esame per i pazienti con sospetta anemia aplastica comprende un esame del sangue clinico completo con determinazione del numero di piastrine e reticolociti, una conta del mielogramma e un esame istologico della biopsia del midollo osseo. Al fine di identificare le varianti della malattia associate alla presenza del clone EPN, si consiglia a tutti i pazienti con anemia aplastica di testare le cellule del sangue per l'umoglobinuria parossistica notturna utilizzando la citometria a flusso altamente sensibile. I potenziali riceventi di midollo osseo vengono sottoposti a tipizzazione HLA delle cellule del sangue.
Per diagnosticare le forme congenite rare della malattia, è importante un'anamnesi approfondita e un esame del paziente. Per escludere l'anemia di Fanconi, è indicata un'analisi cromosomica dei linfociti del sangue, un test per le rotture cromosomiche indotte con diepossibutano o mitomicina.
Effettuando la diagnosi differenziale, è necessario escludere citopenie di origine secondaria. Questo, oltre ad un'anamnesi e ad un esame dettagliati, può richiedere test come la determinazione del livello di vitamina B12 e folati nel sangue, test virali, immunofenotipizzazione delle cellule del midollo osseo, ecografia ed ecocardiografia, test per escludere malattie reumatoidi e altri test come indicato.
La diagnosi differenziale viene effettuata anche con l'aplasia parziale acquisita dei globuli rossi e con la forma congenita dell'anemia di Diamond-Blackfan, in cui viene rilevata l'aplasia della linea eritroide del midollo osseo mentre è preservata la granulo- e trombocitopoiesi.
Classificazione
Per determinare le tattiche di trattamento, è necessario determinare la gravità dell'anemia aplastica. In conformità con la classificazione internazionale, è consuetudine distinguere tra forme gravi e non gravi di anemia aplastica. Lo scopo principale di questa classificazione era identificare un gruppo di pazienti che sono principalmente candidati al trapianto di midollo osseo a causa del rischio di morte prematura.
Trattamento
La strategia di trattamento dell'anemia aplastica dovrebbe mirare a ripristinare la carenza di cellule staminali emopoietiche e a sopprimere i processi immunologici distruttivi.
Il ripristino completo dell'ematopoiesi del midollo osseo nei pazienti con anemia aplastica può essere ottenuto solo con il trapianto di cellule staminali emopoietiche, che rappresenta il metodo di scelta nei pazienti giovani con forme gravi e super-gravi della malattia. Tuttavia, il principale metodo terapeutico per la maggior parte dei pazienti è la terapia immunosoppressiva in quanto metodo più accessibile, con meno controindicazioni e paragonabile in termini di efficacia al trapianto di cellule staminali emopoietiche.
I primi tentativi di trattare l’anemia aplastica con trapianti di midollo osseo furono fatti negli anni ’30, ma la complessità e l’imperfezione della tecnologia di selezione dei donatori e delle tecniche di trapianto dell’epoca limitavano le possibilità di utilizzo del trapianto. Con il miglioramento della tecnologia e dei metodi di selezione dei donatori, il trapianto di midollo osseo è diventato parte del trattamento standard per i pazienti con anemia aplastica grave come metodo di scelta nei pazienti di nuova diagnosi con anemia aplastica grave in presenza di un donatore correlato HLA-identico e come un metodo di trattamento per pazienti con malattia grave che non hanno risposto al trattamento con immunoglobulina antitimocitaria e ciclosporina. Una maggiore efficienza del trapianto allogenico di midollo osseo è stata ottenuta grazie alla riduzione dell’incidenza di complicanze infettive, al miglioramento dei regimi di preparazione pre-trapianto, alla riduzione dell’incidenza delle reazioni di rigetto e della malattia del trapianto contro l’ospite.
Secondo il Gruppo di lavoro europeo sul trapianto di midollo osseo e l’anemia aplastica, il tasso di sopravvivenza dei pazienti con anemia aplastica grave dopo trapianto di cellule staminali ematopoietiche nel 1970-1979 è stato del . 43%, nel 1991-1996 aumentato al 69% e nel periodo 1997-2002. - fino al 72%. La sopravvivenza a lungo termine dei pazienti con anemia aplastica dopo il trapianto può attualmente raggiungere l’80-96%. La fonte preferita di cellule staminali ematopoietiche per i pazienti con anemia aplastica è il midollo osseo.
Nei pazienti con anemia aplastica lieve e anemia aplastica grave che hanno più di 40 anni e/o non hanno un fratello donatore HLA compatibile, si raccomanda un ciclo di terapia immunosoppressiva. L'uso della terapia immunosoppressiva si basa sul concetto della patogenesi dell'anemia aplastica come processo patologico causato da una violazione della regolazione immunitaria dell'ematopoiesi. Il regime standard di terapia immunosoppressiva, che dà i migliori risultati sia per i pazienti con anemia aplastica grave che per anemia aplastica non grave, è una combinazione di immunoglobulina antitimocitaria e ciclosporina A. I vantaggi della terapia di combinazione sono stati confermati da molti gruppi di ricerca. Pertanto, i risultati a 11 anni della terapia immunosoppressiva, secondo un gruppo di scienziati tedeschi, hanno mostrato un aumento della frequenza delle remissioni quando l'immunoglobulina antitimocitaria e la ciclosporina venivano aggiunte alla terapia dal 41 al 70% nel gruppo generale di pazienti e dal 31 al 65% nell'anemia aplastica grave. Allo stesso tempo, il tempo mediano per ottenere la remissione è diminuito da 82 a 60 giorni e l’incidenza senza recidive è aumentata del 18%.
L'immunoglobulina antitimocitaria è un farmaco ottenuto immunizzando gli animali con linfociti umani (timociti fetali). I farmaci di questa serie hanno un effetto linfocitotossico selettivo contro i soppressori T attivati, inibiscono la produzione di citochine soppressive da parte delle cellule T e agiscono sull'apoptosi riducendo l'espressione dell'antigene Fas sulle cellule CD+ del midollo osseo dei pazienti.
La ciclosporina A è un metabolita del fungo Tolipocladium inflatum, un polipeptide ciclico che modifica selettivamente e reversibilmente la funzione dei linfociti, sopprimendo la produzione e la fissazione delle linfochine su recettori specifici; inibisce le fasi G0 e G1 del ciclo cellulare delle cellule immunocompetenti, riduce l'attività dei geni responsabili della sintesi di IL-2 e di una serie di altre citochine. Il vantaggio della CsA è il suo effetto reversibile specifico in assenza di un effetto soppressivo sull’ematopoiesi, nonché la relativa conservazione dell’immunità antinfettiva.
I cicli di terapia con immunoglobulina antitimocitaria, della durata di 4-5 giorni, vengono effettuati in ospedale. Le dosi raccomandate del farmaco per l'immunoglobulina antitimocitaria equina sono 20-40 mg/kg di peso corporeo. Per migliorare i risultati e prevenire reazioni allergiche e malattia da siero, i glucocorticoidi vengono solitamente prescritti contemporaneamente sotto forma di un ciclo breve [metilprednisolone alla dose di 1-3 mg/kg)]. Dopo aver completato la somministrazione di immunoglobulina antitimocitaria, i preparati orali di CsA vengono prescritti per un lungo periodo (da 6 mesi) in dosi di 5-7 mg/kg e superiori in assenza di tossicità significativa. Quando si utilizza questo regime, il tasso di risposta è del 60-80%, con un tasso di sopravvivenza a 5 anni dei pazienti con anemia aplastica grave del 75-85%.
I primi risultati positivi persistenti durante la terapia immunosoppressiva si osservano solitamente dopo 2-3 mesi, pertanto è consigliabile determinare i risultati della terapia dopo 3-6 mesi dall'inizio del trattamento. I criteri per l'efficacia della terapia sono la remissione completa e parziale. La completa remissione clinica ed ematologica presuppone l'assenza dei sintomi clinici della malattia, il completo sollievo della sindrome emorragica e un contenuto di emoglobina superiore a 110 g/l; contenuto di granulociti superiore a 1,0x109/l, contenuto piastrinico superiore a 100x109/l (in altri casi - superiore a 125-150x109/l). La remissione clinica ed ematologica parziale è caratterizzata dall'assenza di sintomi clinici della malattia e manifestazioni di sindrome emorragica, un contenuto di emoglobina superiore a 80 g/l con indipendenza dalla terapia con emocomponenti, un contenuto di granulociti superiore a 0,5x109/l, piastrine più di 20,0x109/l.
Un risultato positivo può anche essere un miglioramento clinico ed ematologico, in cui non si verificano manifestazioni emorragiche pronunciate, la necessità di terapia con emocomponenti è ridotta e si osserva un miglioramento dei parametri ematologici con un contenuto di granulociti superiore a 0,5x109/l, piastrine superiori a 20,0x109/l.
Per valutare l'efficacia del trattamento per i pazienti con anemia aplastica a seconda della gravità della malattia, il Gruppo europeo di esperti propone i seguenti criteri. Secondo le moderne raccomandazioni, la CsA dovrebbe essere continuata dopo aver ottenuto la massima risposta ematologica [remissione parziale sostenuta con miglioramento di tutte le linee ematopoietiche, remissione completa] per 6-12 mesi, seguita da una sospensione graduale, che riduce al minimo il numero di recidive.
Esiste un'esperienza positiva con l'uso di dosi elevate di ciclofosfamide nella prima linea terapeutica. Le prime pubblicazioni risalenti al 1996 mostravano un buon effetto della terapia immunosoppressiva con questi farmaci in pazienti affetti da anemia aplastica, ma con gravi complicanze durante la terapia, comprese infezioni mortali. Tuttavia, con il miglioramento della terapia di accompagnamento, pubblicazioni più recenti mostrano buoni risultati terapeutici con remissioni più complete e durature ottenute in pazienti con anemia aplastica grave, sebbene questi risultati non siano stati confermati da studi randomizzati e controllati.
Se il primo ciclo di terapia di combinazione con immunoglobulina antitimocitaria/CsA è inefficace nei pazienti con anemia aplastica grave, viene presa in considerazione la possibilità di trapianto di midollo osseo da un donatore compatibile non consanguineo. Inoltre, la probabilità di risultati favorevoli è maggiore quando il trapianto viene eseguito in una data precedente.
Gli svantaggi della terapia immunosoppressiva come metodo di trattamento dei pazienti con anemia aplastica includono:
conservazione dei difetti ematopoietici residui (sotto forma di conservazione dei focolai di ipoplasia del midollo osseo, inferiorità funzionale dei mielocariociti);
alto rischio di recidiva (fino al 20-30% dei pazienti e oltre);
complicanze clonali tardive (fino al 20-60% con osservazione a lungo termine), inclusa la sindrome mielodisplastica, la leucemia acuta, l'emoglobinuria parossistica notturna.
La frequenza delle recidive nei pazienti con anemia aplastica dopo la prima linea di terapia immunosoppressiva è relativamente alta, ma nella maggior parte dei casi tali recidive vengono trattate con successo con cicli ripetuti di terapia immunosoppressiva e non peggiorano significativamente la prognosi complessiva. Pertanto, gli studi degli ultimi anni hanno dimostrato che in caso di recidiva dopo il primo ciclo terapeutico di successo, che comprendeva immunoglobulina antitimocitaria, cicli ripetuti portano a remissioni nell'11-65% dei pazienti.
Nella 2a e nelle successive linee terapeutiche è possibile utilizzare farmaci come alemtuzumab, preparati di acido micofenolico per l'intolleranza alla CsA. Esistono prove di esperienze positive con l'uso del farmaco daclizumab (anticorpi monoclonali ricombinanti contro il recettore IL-2) e di una serie di altri farmaci immunosoppressori, ma non esistono ancora dati sufficientemente convincenti sul loro utilizzo in ampi gruppi di pazienti con aplastica. anemia.
La splenectomia, precedentemente utilizzata nel trattamento dei pazienti con anemia aplastica, viene oggi utilizzata raramente, anche se alcuni autori ne ritengono giustificato l'utilizzo nella 2a-3a linea terapeutica, soprattutto in presenza di una componente autoimmune.
È stato dimostrato che l’inizio precoce di un ciclo di trattamento e un’adeguata terapia di accompagnamento sono di grande importanza per migliorare i risultati della terapia per l’anemia aplastica. Quest'ultima comprende la terapia sostitutiva del sangue per mantenere il livello di globuli rossi e piastrine a un livello sicuro.
Le indicazioni per l'uso del concentrato piastrinico sono la sindrome emorragica con livelli piastrinici
Negli ultimi anni sono stati fatti tentativi di utilizzare gli agonisti dei recettori della trombopoietina (eltrombopag) per controllare la sindrome emorragica, che hanno mostrato buoni risultati. Inoltre, esistono dati che indicano la capacità degli agonisti dei recettori della trombopoietina di portare non solo ad un aumento del numero delle piastrine e alla riduzione delle manifestazioni emorragiche, ma anche al miglioramento di altre linee cellulari.
Poiché la dipendenza dalle trasfusioni spesso porta a un sovraccarico di ferro post-trasfusionale nei pazienti con anemia aplastica, i pazienti con frequenti trasfusioni di globuli rossi e livelli di ferritina sierica superiori a 1000 mg/L sono indicati per la terapia chelante del ferro.
Se si verificano complicanze infettive in pazienti con anemia aplastica, la terapia viene effettuata secondo le regole comuni ai pazienti in terapia immunosoppressiva, con la prescrizione di farmaci antibatterici ad ampio spettro e farmaci antifungini secondo indicazione.
La maggior parte dei ricercatori non ritiene consigliabile l'uso di stimolanti ematopoietici - fattore stimolante le colonie di granulociti ed eritropoietina - nei pazienti con anemia aplastica a causa della loro scarsa efficacia in questo gruppo di pazienti e dell'aumento del rischio di sviluppare complicanze clonali. I dati provenienti da osservazioni a lungo termine e meta-analisi, regolarmente presentati negli anni 2000 ai congressi scientifici e alle conferenze di ematologia dell'American Society of Hematology, dell'European Hematology Association, dell'European Group of Bone Marrow Transplantation e altri, hanno dimostrato che l'uso dell'eritropoietina e il G-CSF non influisce in modo significativo sulla riduzione della mortalità o sull'aumento delle risposte complete e complessive alla terapia. Tuttavia, brevi cicli di G-CSF possono essere raccomandati in caso di infezioni sistemiche gravi in pazienti con granulocitopenia profonda. La prognosi della malattia dipende principalmente dalla gravità dell'aplasia e dall'inizio precoce della terapia attiva. Senza trattamento, nelle forme gravi, fino al 50% dei pazienti muore nei primi mesi e con la terapia moderna la sopravvivenza a lungo termine è del 70-80%.
Per quanto riguarda la terapia immunosoppressiva, i risultati del trattamento sono migliori nei pazienti con una risposta granulocitaria e reticolocitaria precoce. I dati disponibili indicano anche una migliore risposta alla terapia immunosoppressiva nei pazienti con anemia aplastica associata alla presenza del clone EPN. Tra i fattori che influenzano la prognosi della malattia vi sono l'efficacia della terapia immunosoppressiva e la probabilità di evoluzione clonale; recentemente l'attenzione è stata attirata dall'accorciamento della lunghezza dei telomeri nelle cellule del sangue.
- In contatto con 0
- Google+ 0
- OK 0
- Facebook 0






