Policitemia vera (eritremia, malattia di Wakez, policitemia eritema) - PV - una malattia mieloproliferativa neoplastica cronica con danno alle cellule staminali, proliferazione di tre linee ematopoietiche, aumento della produzione di globuli rossi e, in misura minore, di globuli bianchi e piastrine. Ad un certo stadio della malattia si unisce la metaplasia mieloide della milza.
L'incidenza della policitemia vera è di circa 1 caso ogni 100.000 abitanti all'anno e ha una chiara tendenza all'aumento negli ultimi anni. Gli uomini si ammalano un po’ più spesso delle donne (1,2:1). L'età media dei pazienti è di 60 anni, i pazienti sotto i 40 anni rappresentano solo il 5%.
Eziopatogenesi. La policitemia vera è una malattia neoplastica clonale, che si basa sulla trasformazione della cellula staminale emopoietica. Poiché la trasformazione maligna avviene a livello di una cellula staminale pluripotente, tutte e tre le linee ematopoietiche sono coinvolte nel processo. Nei pazienti affetti da PV, si riscontra un aumento del contenuto di CFU-GEMM (unità formanti colonie - granulocitiche, eritroidi, macrofagiche e megacariocitiche) - cellule precursori vicine alla cellula staminale pluripotente. Nella coltura cellulare, queste cellule proliferano attivamente in assenza di eritropoietina. Un basso livello di eritropoietina sierica è una caratteristica specifica della PV. Nel midollo osseo si osserva iperplasia prevalentemente di cellule eritroidi, nonché germogli granulocitici e megacariocitici. Una caratteristica caratteristica è la presenza di grappoli di megacariociti polimorfici (da piccoli a giganti). La mielofibrosi si osserva raramente al momento della diagnosi, ma si manifesta distintamente con un lungo decorso della malattia. A poco a poco si osserva un aumento del numero di fibre di reticolina e di collagene, si sviluppa mielofibrosi e la mielopoiesi si riduce. Aumenta la massa degli eritrociti circolanti (MCE), aumenta l'ematocrito, aumenta la viscosità del sangue (si osserva un aumento significativo del contenuto di emoglobina nel sangue (da 180 g / le oltre), eritrociti (da 6,6 x 10 12 / l) e l'indice ematocrito (dal 55% in su). Questi fattori, insieme alla trombocitosi, portano a disturbi della microcircolazione e complicazioni tromboemboliche. Parallelamente si forma metaplasia mieloide della milza. Nel PV non esiste un marcatore citogenetico specifico; anomalie.
Quadro clinico cambia con il decorso della malattia ed è determinata principalmente dallo stadio della malattia. Nella letteratura nazionale è consuetudine distinguere quattro stadi del PV, che riflettono i processi patologici che si verificano nel midollo osseo e nella milza dei pazienti.
Fasi:
I - iniziale, asintomatico (5 anni o più):
la milza non è palpabile
eritrocitosi moderata
pletora moderata
panmielosi nel midollo osseo
le complicanze vascolari e trombotiche sono possibili ma non comuni
Manifestazioni esterne della malattia: pletora, acrocianosi, eritromelalgia (dolore bruciante, parestesia sulla punta delle dita) e prurito della pelle dopo il lavaggio. Un aumento dell'MCE e, di conseguenza, del volume del sangue circolante porta all'ipertensione arteriosa. Se il paziente ha già sofferto di ipertensione, si verifica un aumento della pressione sanguigna e la terapia antipertensiva diventa inefficace. Le manifestazioni della malattia coronarica, dell'aterosclerosi cerebrale sono aggravate. Poiché l'MCE aumenta gradualmente, la pletora, l'aumento del numero degli eritrociti e dell'emoglobina, segni di un disturbo del microcircolo in un certo numero di pazienti compaiono 2-4 anni prima della diagnosi.
II - eritremico, dispiegato (10-15 anni):
A. Senza metaplasia mieloide della milza
la condizione generale è disturbata
pletora grave (Hb 200 g/l o più)
complicanze trombotiche (ictus, infarto miocardico, necrosi dei polpastrelli)
panmielosi
eritromelalgia (dolore agli arti e alle ossa)
Nel quadro del sangue periferico, oltre all'eritrocitosi, è spesso presente la neutrofilia con uno spostamento della formula dei leucociti a sinistra verso singoli mielociti, nonché la basofilia e la trombocitosi. Nel midollo osseo viene rilevata un'iperplasia totale a tre crescita con megacariocitosi pronunciata ed è possibile la mielofibrosi della reticolina. Ma in questo stadio della malattia, la metaplasia mieloide della milza (MMS) è ancora assente e la splenomegalia osservata è dovuta all'aumento del sequestro di eritrociti e piastrine. Le complicanze vascolari sono più frequenti e gravi rispetto al primo stadio della malattia. Nella patogenesi della trombosi, un ruolo importante è giocato dall'aumento dell'MCE, che porta ad un aumento della viscosità del sangue e ad un rallentamento del flusso sanguigno, trombocitosi e disfunzione endoteliale. L'ischemia associata ad un alterato flusso sanguigno arterioso si verifica nel 24-43% dei pazienti. Predominano la trombosi dei vasi cerebrali, gli organi coronari e che forniscono sangue delle arterie della cavità addominale. La trombosi venosa si verifica nel 25-30% dei pazienti ed è la causa di morte in circa un terzo dei pazienti con PV. Trombosi frequente delle vene del sistema portale e delle vene mesenteriche. In un certo numero di pazienti, sono le complicanze trombotiche a diventare una manifestazione del PV. La policitemia vera può essere accompagnata dalla sindrome emorragica: frequenti epistassi e sanguinamento dopo l'estrazione del dente. L'ipocoagulazione si basa su un rallentamento della conversione del fibrinogeno in fibrina, che si verifica in proporzione ad un aumento dell'ematocrito e ad una violazione della retrazione del coagulo sanguigno. Le erosioni e le ulcere dello stomaco e del duodeno sono considerate complicanze viscerali della PV.
B. Con metaplasia mieloide della milza (MMS).
epatosplenomegalia
la pletora è moderatamente espressa
panmielosi
aumento del sanguinamento
complicanze trombotiche
La splenomegalia aumenta, il numero dei leucociti aumenta, lo spostamento della formula dei leucociti a sinistra diventa più pronunciato. Nel midollo osseo - panmielosi; sviluppa gradualmente la reticolina e la mielofibrosi focale del collagene. Il numero di eritrociti e piastrine è leggermente ridotto a causa della loro maggiore distruzione nella milza, nonché della graduale sostituzione del tessuto ematopoietico con tessuto fibroso. In questa fase si può osservare la stabilizzazione delle condizioni dei pazienti, il livello di emoglobina, eritrociti e piastrine si avvicina alla norma senza misure terapeutiche.
III - anemico:
anemico sm (anche pancitopenia)
mielofibrosi pronunciata
fegato, milza ingrossata
Nel midollo osseo aumenta la mielofibrosi del collagene e diminuisce la mielopoiesi. L'emocromo mostra anemia, trombocitopenia, pancitopenia. Nel quadro clinico della malattia possono essere presenti sindromi anemiche ed emorragiche, sono in aumento la splenomegalia e la cachessia. L'esito della malattia può essere la trasformazione in leucemia acuta e sindrome mielodisplastica (MDS).
Diagnostica. Attualmente, per stabilire la diagnosi di policitemia vera vengono utilizzati i criteri sviluppati dall’American Polycythemia Vera Study Group (PVSG). Voi-
1) un aumento della massa degli eritrociti circolanti (più di 36 ml/kg per gli uomini e più di 32 ml/kg per le donne);
2) normale saturazione del sangue arterioso con ossigeno (pO2 superiore al 92%);
3) splenomegalia.
1) trombocitosi (conta piastrinica superiore a 400 x 10 9 /l);
2) leucocitosi (il numero di leucociti è superiore a 12 x 10 9 / senza segni di infezione);
3) attività della fosfatasi alcalina (neutrofili superiori a 100 unità in assenza di febbre o infezione);
4) alto contenuto di vitamina B12 (più di 900 pg/ml).
La diagnosi di PV è considerata affidabile se il paziente presenta tutti e tre i segni della categoria A, oppure se sono presenti il primo e il secondo segno della categoria A e due segni qualsiasi della categoria B.
Attualmente il caratteristico quadro istologico del midollo osseo è considerato il segno diagnostico più importante; iperplasia delle cellule dei germogli eritroidi, granulocitici e megacariocitici con predominanza di eritroidi, accumuli di megacariociti polimorfici (da piccoli a giganti). La mielofibrosi viene osservata raramente al momento della diagnosi, ma si distingue con il lungo decorso della malattia.
Nello stadio I, la policitemia vera, caratterizzata da eritrocitosi isolata, deve essere differenziata dall'eritrocitosi secondaria, che è una risposta a qualsiasi processo patologico nel corpo e può essere sia vera che relativa.
L'eritrocitosi relativa è una conseguenza dell'emoconcentrazione, cioè l'MCE è normale, ma il volume plasmatico è ridotto, cosa che si osserva quando il corpo è disidratato (ad esempio, assumendo diuretici, poliuria nei pazienti con diabete mellito, vomito e diarrea), perdita di una grande quantità di plasma durante le ustioni.
La vera eritrocitosi secondaria (aumento dell'MCE, aumento dell'ematocrito) è dovuta all'aumento della produzione di eritropoietina. Quest'ultimo è di natura compensatoria ed è causato dall'ipossia tissutale nelle persone che vivono a notevole altezza sul livello del mare, nei pazienti con patologie del sistema cardiovascolare e respiratorio e nei fumatori. Questa categoria comprende anche pazienti con emoglobinopatie ereditarie, caratterizzate da una maggiore affinità dell'emoglobina per l'ossigeno, che viene rilasciato nei tessuti del corpo in quantità minore. Una produzione inadeguata di eritropoietina si osserva nelle malattie renali (idronefrosi, patologia vascolare, cisti, tumori, anomalie congenite), carcinoma epatocellulare, grande mioma uterino. Un segno diagnostico differenziale essenziale è il livello di eritropoietina nel siero del sangue.
Trattamento. Nelle fasi iniziali della malattia si consiglia di utilizzare il salasso, che allevia notevolmente le manifestazioni della sindrome pletorica. Il metodo di scelta per abbassare l'ematocrito (e l'emoglobina a valori normali) è il salasso (esfusione), consigliato se l'ematocrito supera 0,54. L'obiettivo del trattamento è un ematocrito inferiore a 0,42 per le donne e 0,45 per gli uomini. Nelle condizioni moderne, il salasso può essere sostituito dall'eritrocitoaferesi. Inoltre, per facilitare il salasso e prevenire complicazioni trombotiche, ai pazienti vengono somministrati cicli di terapia antipiastrinica (aspirina, reopoligliukina, ecc.). La scelta del metodo di trattamento nello stadio II avanzato della PV è forse il compito più difficile. Oltre all'eritrocitosi, i pazienti presentano leucocitosi e trombocitosi e queste ultime possono raggiungere numeri molto elevati. Alcuni pazienti hanno già manifestato complicazioni trombotiche e le efusioni aumentano il rischio di trombosi.
Quando si individualizza la terapia, è necessario tenere conto dell’età dei pazienti. Pertanto il trattamento dei pazienti di età inferiore ai 50 anni, senza storia di complicanze trombotiche e grave ipertrombocitosi (< 1000,0 х 10 9 /л) может быть ограничено только кровопусканиями в сочетании с терапией аспирином (или без него) в дозе 100-375 мг в день.
I pazienti di età superiore a 70 anni con una storia di complicanze trombotiche e grave ipertrombocitosi vengono trattati con farmaci mielosoppressivi. I pazienti di età compresa tra 50 e 70 anni senza complicanze trombotiche e grave ipertrombocitosi possono essere trattati con agenti mielosoppressori o salasso, sebbene quest'ultimo possa aumentare il rischio di complicanze trombotiche.
Attualmente, oltre ai salassi e agli agenti antipiastrinici, per il trattamento del PV vengono utilizzati principalmente l'idrossiurea e l'interferone alfa, meno spesso il busulfan e all'estero viene utilizzato l'anagrelide. L'idrossiurea può essere il farmaco di scelta se i pazienti con PV presentano leucocitosi e trombocitosi gravi. Ma per i pazienti giovani, l'uso dell'idrossiurea è limitato dai suoi effetti mutageni e leucosogenici. Oltre all'idrossiurea, l'interferone alfa è ampiamente utilizzato nel trattamento del PV. In primo luogo, l'IF-a sopprime bene la proliferazione patologica e non ha un effetto leucemogenico. In secondo luogo, come l'idrossiurea, riduce significativamente la produzione di piastrine e leucociti. Particolarmente degna di nota è la capacità di IF-a di eliminare il prurito causato dall'assunzione di procedure idriche.
L'aspirina in una dose giornaliera di 50-250 mg, di regola, elimina i disturbi della microcircolazione. L'uso di questo farmaco o di altri agenti antipiastrinici a scopo terapeutico o profilattico è raccomandato per tutti i pazienti con PV.
Sfortunatamente, attualmente non esiste un trattamento efficace per il PV anemico di stadio III. La terapia è limitata alle cure palliative. La sindrome anemica ed emorragica viene corretta mediante trasfusioni di componenti del sangue. È stata segnalata l'efficacia del trapianto di cellule staminali emopoietiche in pazienti con PV in fase di mielofibrosi con splenomegalia e pancitopenia e trasformazione in leucemia acuta o MDS. La sopravvivenza a tre anni dei pazienti dopo il trapianto è stata del 64%.
Previsione. Nonostante un decorso lungo e in alcuni casi favorevole, la PV è una malattia grave ed è irta di complicazioni fatali che riducono l'aspettativa di vita dei pazienti. La causa più comune di morte nei pazienti è la trombosi e l'embolia (30-40%). Nel 20-50% dei pazienti nello stadio di mielofibrosi post-policitemica (stadio III PV), si verifica una trasformazione in leucemia acuta, che ha una prognosi sfavorevole - un tasso di sopravvivenza a tre anni solo del 30%.
- emoblastosi cronica, che si basa sulla proliferazione illimitata di tutti i germogli della mielopoiesi, principalmente degli eritrociti. Clinicamente, la policitemia si manifesta con sintomi cerebrali (pesantezza alla testa, vertigini, acufeni), sindrome tromboemorragica (trombosi arteriosa e venosa, sanguinamento), disturbi microcircolatori (freddo delle estremità, eritromelalgia, iperemia della pelle e delle mucose). Le principali informazioni diagnostiche si ottengono dallo studio del sangue periferico e del midollo osseo. Per trattare la policitemia vengono utilizzati salassi, eritrocitaferesi e chemioterapia.

informazioni generali
Cause della policitemia
Lo sviluppo della policitemia è preceduto da cambiamenti mutazionali nella cellula ematopoietica staminale pluripotente, che dà origine a tutte e tre le linee cellulari del midollo osseo. La mutazione rilevata più frequentemente del gene della tirosina chinasi JAK2 con la sostituzione della valina con fenilalanina in posizione 617. A volte si riscontra un'incidenza familiare di eritremia, ad esempio tra gli ebrei, il che può indicare a favore di una correlazione genetica.
Con la policitemia nel midollo osseo ci sono 2 tipi di cellule precursori dell'ematopoiesi eritroide: alcune di esse si comportano in modo autonomo, la loro proliferazione non è regolata dall'eritropoietina; altri, come previsto, sono eritropoietina-dipendenti. Si ritiene che una popolazione cellulare autonoma non sia altro che un clone mutante, il principale substrato della policitemia.
Patogenesi
Nella patogenesi dell'eritremia, il ruolo principale appartiene all'aumento dell'eritropoiesi, che provoca eritrocitosi assoluta, alterazioni delle proprietà reologiche e di coagulazione del sangue e metaplasia mieloide della milza e del fegato. L'elevata viscosità del sangue provoca una tendenza alla trombosi vascolare e al danno tissutale ipossico, mentre l'ipervolemia provoca un aumento del riempimento sanguigno degli organi interni. Alla fine della policitemia si notano deplezione dell'ematopoiesi e mielofibrosi.
Classificazione
In ematologia esistono 2 forme di policitemia: vera e relativa. La policitemia relativa si sviluppa con un livello normale di eritrociti e una diminuzione del volume plasmatico. Questa condizione è chiamata stress o falsa policitemia e non è considerata nell'ambito di questo articolo.
La vera policitemia (eritremia) per origine può essere primaria e secondaria. La forma primaria è una malattia mieloproliferativa indipendente, che si basa sulla sconfitta del germe mieloide dell'ematopoiesi.
La policitemia secondaria di solito si sviluppa con un aumento dell'attività dell'eritropoietina; questa condizione è una reazione compensatoria all'ipossia generale e può verificarsi con patologia polmonare cronica, difetti cardiaci "blu", tumori surrenalici, emoglobinopatie, quando si sale in altezza o si fuma, ecc. La vera policitemia nel suo sviluppo attraversa 3 fasi: iniziale, avanzato e terminale.
Metto in scena(iniziale, oligosintomatico) - dura circa 5 anni; asintomatici o con manifestazioni cliniche minime. È caratterizzata da moderata ipervolemia, lieve eritrocitosi; la dimensione della milza è normale.
II stadio(eritremico, espanso) si divide in due sottostadi:
- IA - senza trasformazione mieloide della milza. Sono presenti eritrocitosi, trombocitosi, talvolta pancitosi; secondo il mielogramma - iperplasia di tutti i germogli ematopoietici, megacariocitosi pronunciata. La durata dello stadio avanzato dell'eritremia è di 10-20 anni.
- IIB - con presenza di metaplasia mieloide della milza. Sono espressi ipervolemia, epato e splenomegalia; nel sangue periferico - pancitosi.
III stadio(anemico, posteritremico, terminale). Caratterizzato da anemia, trombocitopenia, leucopenia, trasformazione mieloide del fegato e della milza, mielofibrosi secondaria. Possibili esiti della policitemia in altre emoblastosi.
Sintomi della policitemia
L'eritremia si sviluppa a lungo, gradualmente e può essere rilevata per caso durante un esame del sangue. I primi sintomi, come pesantezza alla testa, acufeni, vertigini, visione offuscata, brividi alle estremità, disturbi del sonno, ecc., vengono spesso "attribuiti" all'età avanzata o a malattie concomitanti.
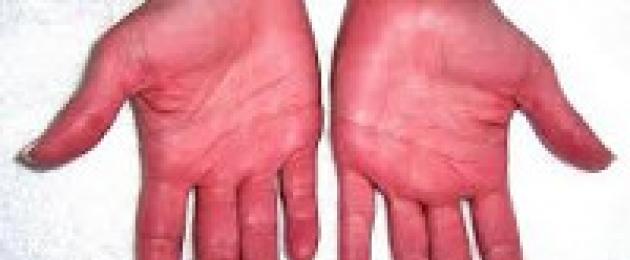
La caratteristica più caratteristica della policitemia è lo sviluppo di una sindrome pletorica causata da pancitosi e un aumento del BCC. La telangiectasia, la colorazione rosso ciliegia della pelle (soprattutto viso, collo, mani e altre aree aperte) e delle mucose (labbra, lingua), l'iperemia della sclera servono come prova della pletora. Un tipico segno diagnostico è il sintomo di Cooperman: il colore del palato duro rimane normale e il palato molle acquisisce una tonalità cianotica stagnante.
Un altro sintomo distintivo della policitemia è il prurito, aggravato dopo le procedure idriche e talvolta insopportabile. Tra le manifestazioni specifiche della policitemia c'è anche l'eritromelalgia, una dolorosa sensazione di bruciore alla punta delle dita, accompagnata dalla loro iperemia.
Nello stadio avanzato dell'eritremia possono manifestarsi emicranie lancinanti, dolori ossei, cardialgia, ipertensione arteriosa. L'80% dei pazienti presenta splenomegalia moderata o grave; il fegato aumenta un po' meno spesso. Molti pazienti con policitemia notano un aumento del sanguinamento delle gengive, lividi sulla pelle, sanguinamento prolungato dopo l'estrazione del dente.
La conseguenza dell'eritropoiesi inefficace nella policitemia è un aumento della sintesi dell'acido urico e una violazione del metabolismo delle purine. Ciò trova espressione clinica nello sviluppo della cosiddetta diatesi uratica: gotta, urolitiasi, colica renale.
Complicazioni
Il risultato della microtrombosi e della violazione del trofismo della pelle e delle mucose sono ulcere trofiche della parte inferiore della gamba, ulcere gastriche e duodenali. Le complicanze più frequenti nella clinica della policitemia sono la trombosi vascolare delle vene profonde, dei vasi mesenterici, delle vene porta, delle arterie cerebrali e coronarie. Le complicanze trombotiche (EP, ictus ischemico, infarto miocardico) sono le principali cause di morte nei pazienti con policitemia. Allo stesso tempo, insieme alla trombosi, i pazienti con policitemia sono inclini alla sindrome emorragica con lo sviluppo di sanguinamento spontaneo di varie localizzazioni (gengivale, nasale, dalle vene dell'esofago, gastrointestinale, ecc.).
Diagnostica
I cambiamenti ematologici che caratterizzano la policitemia sono decisivi nella diagnosi. Un esame del sangue rivela eritrocitosi (fino a 6,5-7,5x10 12 / l), aumento dell'emoglobina (fino a 180-240 g / l), leucocitosi (oltre 12x10 9 / l), trombocitosi (oltre 400x10 9 / l). La morfologia degli eritrociti, di regola, non è cambiata; con un aumento del sanguinamento, può essere rilevata la microcitosi. Una conferma affidabile dell'eritremia è un aumento della massa degli eritrociti circolanti superiore a 32-36 ml / kg.
Per lo studio del midollo osseo nella policitemia, è più informativo eseguire non una puntura sternale, ma una trepanobiopsia. L'esame istologico della biopsia ha rivelato panmielosi (iperplasia di tutti i germogli emopoietici), negli stadi successivi della policitemia - mielofibrosi secondaria.
Per valutare il rischio di sviluppare complicanze dell'eritremia, ulteriori esami di laboratorio (esami funzionali del fegato, analisi generale delle urine) e studi strumentali (ecografia dei reni, ecografia delle vene delle estremità, ecocardiografia, ecografia dei vasi della testa e del collo , FGDS, ecc.) vengono eseguiti. Con la minaccia di disturbi tromboemorragici e metabolici, sono necessarie consultazioni con specialisti ristretti pertinenti: un neurologo, un cardiologo, un gastroenterologo e un urologo.
Trattamento della policitemia
Per normalizzare il volume del BCC e ridurre il rischio di complicanze trombotiche, il salasso è la prima misura. Le estrazioni di sangue vengono effettuate in un volume di 200-500 ml 2-3 volte a settimana, seguite dal rifornimento del volume di sangue rimosso con soluzione salina o reopoliglucina. La conseguenza di frequenti salassi può essere lo sviluppo di anemia da carenza di ferro. Il salasso nella policitemia può essere sostituito con successo dall'eritrocitoferesi, che consente di rimuovere solo la massa di globuli rossi dal flusso sanguigno, restituendo il plasma.
In caso di pronunciati cambiamenti clinici ed ematologici, sviluppo di complicanze vascolari e viscerali, ricorrono alla terapia mielosoppressiva con citostatici (busulfan, mitobronitolo, ciclofosfamide, ecc.). A volte viene eseguita la terapia con fosforo radioattivo. Per normalizzare lo stato aggregato del sangue, vengono prescritti eparina, acido acetilsalicilico, dipiridamolo sotto il controllo di un coagulogramma; in caso di emorragie vengono mostrate trasfusioni di piastrine; con diatesi uratica - allopurinolo.
Previsione
Il decorso dell'eritremia è progressivo; la malattia non è soggetta a remissioni spontanee e guarigione spontanea. I pazienti sono costretti a essere sotto la supervisione di un ematologo per tutta la vita, a sottoporsi a cicli di terapia con emoexfusione. Con la policitemia, il rischio di complicanze tromboemboliche ed emorragiche è elevato. L'incidenza della trasformazione della policitemia in leucemia è dell'1% nei pazienti che non hanno ricevuto chemioterapia e dell'11-15% in quelli trattati con terapia citostatica.
Lettura 8 minuti. Visualizzazioni 144
L'eritremia è una malattia di natura tumorale, in cui si verifica un progressivo aumento del contenuto dei globuli rossi nel sangue. I pazienti affetti da questa malattia necessitano di un trattamento tempestivo, poiché la sua mancanza può portare alla morte.

Cause e classificazione
La causa esatta della malattia di Wakez non è stata ancora stabilita. Tuttavia, ci sono una serie di fattori che aumentano il rischio di svilupparlo. Questi includono:
- predisposizione genetica. La malattia dell'eritremia del sangue si riscontra spesso in persone con patologie genetiche come la sindrome di Down, la sindrome di Klinefelter, la sindrome di Marfan, la sindrome di Bloom.
- Esposizione a sostanze tossiche. Le tossine sono in grado di penetrare nel sangue e causare lo sviluppo di mutazioni. I mutageni chimici includono agenti antibatterici (cloramfenicolo), citostatici, benzene.
- Radiazione ionizzante. Le radiazioni vengono parzialmente assorbite dalle cellule del corpo umano, causando tutti i tipi di disturbi. A rischio sono i residenti in zone sfavorevoli (dal punto di vista ambientale) e i pazienti sottoposti a radioterapia per la presenza di neoplasie maligne.
La patologia è un tipo di leucemia. La classificazione della malattia si basa sui seguenti fattori:
- forma di perdita: acuta, cronica;
- tipo di sviluppo: vero, relativo (falso);
- meccanismo di generazione: primario, secondario.
Fasi e loro sintomi
Nelle prime fasi dello sviluppo, la malattia non si rileva in alcun modo, quindi la persona non è a conoscenza della sua presenza. Man mano che progredisce, si verificano i primi sintomi di eritremia.
Iniziale
Sintomi della malattia allo stadio 1:
Quanto spesso fai un esame del sangue?
Le opzioni del sondaggio sono limitate perché JavaScript è disabilitato nel tuo browser.
Solo su prescrizione del medico curante 30%, 949 voti
Una volta all'anno e penso che sia sufficiente il 18%, 554 votazione
Almeno due volte l'anno 15%, 460 voti
Più di due volte l'anno ma meno di sei volte 11%, 344 votazione
Controllo la mia salute e lo prendo una volta al mese 6%, 197 voti
Ho paura di questa procedura e cerco di non superare il 4%, 135 voti
21.10.2019
- Arrossamento della pelle, delle mucose. La causa dell'iperemia risiede nell'aumento della concentrazione dei globuli rossi nel sangue. La pelle diventa rosa pallido o rossa, inoltre, su tutte le parti del corpo.
- Sensazioni spiacevoli alle dita, agli arti. La presenza di dolore nell'eritremia è dovuta alla ridotta circolazione sanguigna nei piccoli vasi.
Alcuni pazienti possono avvertire mal di testa.
eritremico
Con lo sviluppo della malattia inizia la fase successiva dell'eritremia, i cui sintomi sono più pronunciati. Questi includono:
- Deterioramento del benessere generale. I processi patologici che si verificano nel corpo causano debolezza, vertigini, affaticamento, tinnito e altri sintomi spiacevoli.
- Eritromelalgia. Accompagnato dalla comparsa di macchie viola e dolore bruciante, localizzato sulla punta delle dita delle mani e dei piedi.
- Splenomegalia ed epatomegalia (ingrossamento della milza e del fegato).
- Aumento dell'iperemia della pelle e delle mucose, comparsa di vene gonfie.
- Malattie del tratto gastrointestinale. I processi trofici interferiscono con il normale flusso di sangue ai tessuti, il che porta allo sviluppo di ulcera duodenale e ulcere allo stomaco.
- Sanguinamento (incluso aumento del sanguinamento delle gengive).
- Prurito cutaneo (soprattutto dopo il contatto con l'acqua).
- Forte dolore articolare.
- Aumento della pressione sanguigna.
anemico
Man mano che la malattia di Wakez progredisce, i sintomi peggiorano. Il paziente si trova ad affrontare le seguenti condizioni patologiche:
- Forte sanguinamento. Si verificano spontaneamente o sullo sfondo di lesioni. In alcuni casi, l’emorragia non può essere fermata per diverse ore.
- Anemia. La carenza di ferro è accompagnata da sbiancamento della pelle, deterioramento del benessere generale, comparsa di debolezza e vertigini.
- formazione di trombi. La formazione di placche trombotiche porta a disturbi circolatori nei vasi del cervello, degli arti inferiori, ecc. In questi casi, la probabilità di morte aumenta notevolmente.
Diagnostica
La diagnosi della malattia di Wakez è complessa. Per stabilire il fatto della presenza di patologia, vengono svolte le seguenti attività:
- esame del sangue generale e biochimico;
- ecografia;
- puntura del midollo osseo;
- dopplerografia.
Analisi del sangue
Il primo studio che viene effettuato in relazione ai pazienti con sospetta eritremia è un emocromo completo. Il materiale biologico viene prelevato a stomaco vuoto, in una stanza separata. L'infermiera pulisce la punta del dito con alcol, poi fa una puntura e preleva alcuni millilitri di sangue.

Se una persona soffre di una malattia, ciò viene rilevato da un cambiamento in diversi indicatori. Nelle fasi iniziali dell'eritremia si osserva un aumento dell'emoglobina, del contenuto di eritrociti, piastrine e leucociti. Le fasi tardive sono caratterizzate da una diminuzione delle prestazioni dovuta allo sviluppo dell'anemia.
Biochimica del sangue
Un esame del sangue biochimico viene utilizzato per determinare i seguenti indicatori:
- livello di bilirubina (dipende dall'intensità);
- la quantità di ferro nel sangue;
- contenuto di acido urico;
- il livello dei test epatici (dipende dall'intensità della distruzione delle cellule epatiche).
La policitemia è un processo tumorale in cui aumentano gli elementi cellulari del midollo osseo (iperplasia). La stragrande maggioranza del processo è benigna, sebbene in determinate condizioni sia possibile la transizione verso una forma maligna.
Questa patologia, chiamata eritremia, viene individuata come una forma nosologica separata (malattia). Viene utilizzato anche il nome malattia di Wakez, dal nome del medico che per primo la descrisse nel 1892.
Più spesso la malattia viene diagnosticata negli uomini anziani. Ma per i giovani e la mezza età è caratteristica la predominanza delle donne. La policitemia si manifesta in modi diversi, ma in termini di effetti esterni, le vene della pelle si dilatano e il colore della pelle cambia. I cambiamenti nel collo, sul viso e sulle mani sono particolarmente chiaramente visibili.
La malattia è pericolosa, in particolare trombosi e aumento del sanguinamento (ad esempio dalle gengive).
La policitemia (eritremia, malattia di Wakez, policitemia vera) è una malattia clonale mieloproliferativa neoplastica cronica in cui vi è una proliferazione incontrollata di germogli di mielopoiesi eritroide, megacariocitica e granulocitica con proliferazione predominante di germogli eritroidi (panmielosi), un aumento della concentrazione di eritrociti, un aumento dei livelli di emoglobina, un alto contenuto di piastrine, leucociti (pancitosi).
Importante! Con l'eritremia, l'eritropoiesi non dipende dai normali meccanismi regolatori.
La malattia più comune si verifica negli uomini di mezza età e in età avanzata, ma in generale l'eritremia è una malattia rara.
Per riferimento. La vera policitemia è più comune tra gli ebrei, e gli abitanti più “resistenti” del nostro pianeta a tale malattia sono la razza negroide e gli abitanti del Giappone (l'eccezione sono quelli sopravvissuti agli attacchi atomici).
Eritremia: cancro o no
La policitemia vera appartiene al gruppo delle leucemie croniche, il cui decorso può essere benigno o maligno. Poiché il sistema sanguigno è interessato, questa malattia non può essere chiamata cancro, poiché il cancro è una neoplasia maligna che si sviluppa dai tessuti epiteliali di vari organi.
Tuttavia, l'eritremia è un processo neoplastico altamente differenziato che colpisce il sistema emopoietico umano.
Malattia di Wakez: cause e fattori di rischio
 La causa principale della vera policitemia (primaria) sono le mutazioni genetiche ereditarie, il che è dimostrato dal fatto che quasi tutti i pazienti affetti da questa malattia sono portatori della mutazione JAK2V617F o di altre mutazioni funzionalmente simili.
La causa principale della vera policitemia (primaria) sono le mutazioni genetiche ereditarie, il che è dimostrato dal fatto che quasi tutti i pazienti affetti da questa malattia sono portatori della mutazione JAK2V617F o di altre mutazioni funzionalmente simili.
In tali casi, vengono determinati geni specifici responsabili della sintesi dei globuli rossi e che mostrano un'elevata sensibilità all'eritropoietina. Questo fenomeno è spesso registrato nei parenti ed è familiare.
Un'altra opzione per una mutazione genetica è che i geni patologici inizino a catturare molto ossigeno senza cederlo ai tessuti.
La policitemia secondaria è il risultato di cambiamenti patologici nelle malattie croniche a lungo termine che stimolano la produzione di eritropoietina. Queste malattie e condizioni includono:
- Enfisema dei polmoni.
- Asma bronchiale.
- Bronchite ostruttiva.
- Difetti cardiaci nella fase di compensazione e scompenso.
- Tromboembolia di qualsiasi localizzazione.
- Aumento della pressione nell'arteria polmonare.
- Disturbi del ritmo cardiaco.
- Insufficienza cardiaca.
- Ischemia cardiaca.
- Cisti renali.
- Ischemia renale dovuta a lesioni aterosclerotiche dei vasi renali.
- Tumori del midollo osseo rosso.
- Carcinoma a cellule renali.
- Carcinoma del fegato.
- Processi tumorali nell'utero.
- Tumori delle ghiandole surrenali.
- Fumare.
- Radiazione ionizzante.
- Esposizione a sostanze tossiche e chimiche.
- Alcuni farmaci: cloramfenicolo, azatioprina, metotrexato, ciclofosfamide.
Esistono anche una serie di malattie genetiche che aumentano il rischio di sviluppare la policitemia. Tali malattie non hanno nulla a che fare con il sistema sanguigno, ma l'instabilità genetica porta al fatto che le cellule del sangue diventano più suscettibili a varie influenze esterne ed interne, che possono causare lo sviluppo di eritremia. Tali malattie sono:
- Sindrome di Down.
- Sindrome di Klinefelter.
- Sindrome di fioritura.
- Sindrome di Marfan.
Con la policitemia, la manifestazione principale è un aumento del numero di globuli rossi nel plasma sanguigno, ma le cause di questo processo dipendono direttamente dal tipo di eritremia:
- Tipo assoluto- in questo caso si verifica un aumento della concentrazione degli eritrociti nel sangue a causa della loro maggiore formazione. Questo fenomeno è tipico di:
- Vera policitemia.
- Policitemia in caso di ipossia.
- Ostruzioni polmonari.
- Ipossia che si verifica con danni ai reni, alle ghiandole surrenali.
- Tipo relativo- allo stesso tempo, il volume degli eritrociti aumenta a causa della diminuzione del volume plasmatico. Gli indicatori degli eritrociti non cambiano contemporaneamente, ma cambia il rapporto eritrociti / plasma e quindi questo fenomeno è chiamato relativo. Questo tipo di processo si verifica a causa del verificarsi delle seguenti malattie:
- Salmonellosi.
- Colera.
- Dissenteria e altre malattie infettive accompagnate da vomito e diarrea gravi.
- Brucia.
- Esposizione alle alte temperature, accompagnata da una maggiore sudorazione.
Oltre alle cause immediate dello sviluppo della malattia di Wakez, esistono anche fattori di rischio che, in determinate condizioni, possono innescare il processo patologico:
- Situazioni stressanti, esposizione prolungata allo stress.
- Attività associate alla costante esposizione all'anidride carbonica, che porta a cambiamenti nella composizione del gas nel sangue.
- Vivere negli altopiani per molto tempo.
Come si sviluppa la malattia
I meccanismi patogenetici per lo sviluppo della policitemia si basano su mutazioni nella cellula staminale ematopoietica pluripotente, da cui inizia lo sviluppo del processo patologico:- La mutazione puntiforme V617F si verifica nel gene Jak2, che porta ad un'interruzione nella struttura del gene stesso.
- Di conseguenza, l'attività della tirosina chinasi aumenta in modo significativo, che si trasforma in una maggiore proliferazione delle cellule mature dei germi mieloidi. In questo caso si verifica un blocco completo dell'apoptosi (morte naturale delle cellule).
- Inoltre, una maggiore sensibilità delle cellule precursori patologiche all'eritropoietina, anche alle sue concentrazioni più basse, porta ad un'aumentata sintesi degli elementi formati, in particolare degli eritrociti. Inoltre, esiste anche un secondo tipo di cellule: i precursori degli eritrociti, che si comportano in modo assolutamente indipendente e autonomo, la loro divisione non dipende dall'eritropoietina. Questa popolazione è mutante ed è uno dei principali substrati dell'eritremia.
- Come risultato di tale reazione, si verifica l'iperplasia dei germi ematopoietici con un aumento significativo della produzione di globuli rossi nel midollo osseo in primo luogo, nonché di piastrine e granulociti. Allo stesso tempo si sviluppa l'eritrocitosi assoluta, le proprietà reologiche del sangue sono disturbate.
- Organi e tessuti traboccano di sangue, la cui viscosità aumenta in modo significativo, il che porta allo sviluppo di coaguli di sangue all'interno dei vasi, alterazioni del fegato, milza di varia gravità (metaplasia mieloide della milza e del fegato), ipossia e ipervolemia.
- Nelle fasi finali, l'ematopoiesi si esaurisce, si sviluppa mielofibrosi.
Importante! Un clone cellulare anormale è in grado di trasformarsi in qualsiasi cellula del sangue: eritrociti, leucociti e/o piastrine.
Il risultato di tutte le reazioni patogenetiche è l'emergere di due tipi di cellule - precursori:
- Normale.
- Mutante.
Poiché il processo di formazione delle cellule mutanti è incontrollabile, il numero di eritrociti supera significativamente il fabbisogno del corpo. Ciò porta all'inibizione della sintesi dell'eritropoietina nei reni, che aggrava ulteriormente il processo patologico, poiché l'eritropoietina perde il suo effetto sull'eritropoiesi normale e non ha alcun effetto sulle cellule tumorali.
Inoltre, la crescita costante delle cellule mutanti porta allo spostamento di quelle normali, che ad un certo punto porta al fatto che tutti gli eritrociti sono prodotti da cellule precursori mutanti.
Classificazione delle malattie
Come accennato in precedenza, a seconda dei motivi che hanno portato allo sviluppo della policitemia, questa si divide in due tipologie: 
- Vera policitemia.
- Parente.
La vera eritremia, a sua volta, può essere:
- Primario: la base di questo processo è la sconfitta del germe mieloide dell'ematopoiesi.
- Secondario - la base di questa varietà - un aumento dell'attività dell'eritropoietina.
La malattia attraversa tre fasi di sviluppo:
- Stadio 1 - oligosintomatico, iniziale, altezza - durante questo periodo non ci sono praticamente manifestazioni cliniche di eritremia. Questa fase dura a lungo, fino a 5 anni o più. Durante questo periodo si sviluppano i seguenti processi:
- Ipervolemia moderata.
- eritrocitosi moderata.
- I cambiamenti nella dimensione della milza non vengono rilevati.
- Stadio 2 - dispiegato, eritremico - in questa fase vengono espressi tutti i segni clinici rilevanti. Questo periodo della malattia si divide a sua volta in 2 fasi:
- IA - nessuna degenerazione mieloide della milza. Si sviluppa eritrocitosi, trombocitosi e in alcuni casi pancitosi. Il mielogramma mostra iperplasia di tutte le linee emopoietiche e grave megacariocitosi. Questa fase può durare fino a 20 anni.
- IIB - qui è già coinvolta attivamente la milza, che va incontro a metaplasia mieloide. Si sviluppa una grave ipervolemia, la milza e il fegato aumentano di dimensioni e la pancitosi viene registrata nel plasma sanguigno.
- Stadio 3 - terminale, anemico, posteritremico - lo stadio finale della malattia. Si sviluppa:
- Anemia.
- trombocitopenia.
- Leucopenia.
- Trasformazione mieloide del fegato, milza.
- Mielofibrosi secondaria.
- È possibile degenerare in altre emoblastosi, molto più pericolose della stessa policitemia.
Importante! Nell'ultimo stadio della malattia, le cellule perdono la capacità di differenziarsi, il che nella maggior parte dei casi porta allo sviluppo della leucemia acuta.
Policitemia. Sintomi
Le principali manifestazioni cliniche dell'eritremia sono due sindromi principali:- Pletorico (pletora) I principali sintomi di questa sindrome sono:
- Cambiamento del volume degli eritrociti nel sangue circolante nella direzione dell'aumento.
- Il verificarsi di vertigini, mal di testa.
- Disturbo visivo.
- Sviluppo di prurito alla pelle.
- Angina pectoris.
- La comparsa di una tinta bluastra sulla pelle e sulle mucose visibili, che è chiamata un sintomo positivo di Cooperman.
- Trombosi di qualsiasi livello di localizzazione.
- Arrossamento delle dita degli arti superiori e inferiori, accompagnato da attacchi estremamente dolorosi e una sensazione di bruciore, chiamata eritromelalgia.
- Mieloproliferativo- si verifica a causa dell'iperplasia di tutti e tre i germogli ematopoietici, con i quali sono presenti:
- Sudorazione.
- Prurito sulla pelle.
- Debolezza marcata.
- Aumento della temperatura corporea.
- Violazione del metabolismo delle purine, che causa la diatesi dell'acido urico, la comparsa di calcoli renali, gotta e artrite gottosa.
- Lo sviluppo dell'ematopoiesi extramidollare (i focolai della formazione di cellule del sangue patologiche non compaiono più nel midollo osseo, ma al di fuori di esso).
- Ingrandimento della milza.
- Infezioni frequenti.
Se parliamo di ogni stadio della policitemia, allora sono caratterizzati da segni clinici speciali, che sono segni degli stadi della malattia:
- stato iniziale- non ci sono praticamente manifestazioni qui, non sono specifiche e possono essere attribuite a molte altre malattie di vari organi e sistemi:
- Arrossamento delle mucose e della pelle: questo sintomo si verifica a causa di un aumento della concentrazione dei globuli rossi. Appare in tutte le parti del corpo umano. all'inizio della malattia può essere lieve.
- Mal di testa: si sviluppano in violazione dei processi di microcircolazione nei vasi cerebrali di piccolo calibro.
- Dolore alle dita dei piedi, alle mani - poiché durante questo periodo il flusso sanguigno attraverso i piccoli vasi è già disturbato, ciò porta ad un aumento della viscosità del sangue, che porta ad una diminuzione dell'apporto di ossigeno agli organi. Ciò porta allo sviluppo dell'ischemia e alla comparsa del dolore ischemico.
- Stadio espanso- in questa fase della malattia, la policitemia provoca un aumento significativo del numero delle cellule del sangue, che porta ad un aumento della sua viscosità, alla loro maggiore distruzione nella milza e a disturbi nell'attività del sistema di coagulazione del sangue. Clinicamente, questo si manifesta con tali segni:
- Il rossore della pelle e delle mucose si intensifica fino alla comparsa di una tinta viola e blu.
- Teleaniectasie (emorragie localizzate sulla pelle).
- Si intensifica l'eritromelalgia bilaterale, complicata dalla necrosi delle dita degli arti superiori e inferiori. Un tale processo con la progressione della policitemia può coprire completamente l'intera mano e il piede. Gli attacchi di dolore acuto possono durare fino a diverse ore e l’esposizione all’acqua fredda può fornire un certo sollievo.
- Un aumento del fegato (a volte fino a 10 kg) è espresso dallo sviluppo del dolore nell'ipocondrio destro, un disturbo nell'atto respiratorio e disturbi del processo digestivo.
- Ingrossamento della milza - l'eccessivo riempimento della milza con il sangue porta non solo al suo ingrossamento, ma anche all'ispessimento della milza.
- L'ipertensione arteriosa appare a causa di un aumento del volume del sangue circolante, dell'elevata viscosità del sangue. Ciò provoca lo sviluppo di resistenza vascolare al flusso sanguigno.
- La gravità del prurito cutaneo diventa più forte, questo perché l'aumentata formazione di elementi del sangue, in particolare dei leucociti, porta alle loro elevate concentrazioni. Ciò porta alla loro massiccia distruzione, a seguito della quale l'istamina viene rilasciata attivamente da loro, che è il colpevole del prurito della pelle, che è ulteriormente aggravato dal contatto con l'acqua.
- Aumento del sanguinamento: anche i tagli minori e le lesioni possono portare a sanguinamento a causa dell'elevata pressione sanguigna, dell'aumento del volume del sangue e dell'eccessiva attività piastrinica.
- Lesioni ulcerose del tratto digestivo, che sono accompagnate da sintomi dispeptici di varia gravità.
- Dolore alle articolazioni di qualsiasi localizzazione.
- Ictus ischemico dovuto a trombosi massiva.
- Infarto miocardico.
- Segni di carenza di ferro: unghie esfolianti, pelle secca e mucose, crepe agli angoli della bocca, scarso appetito, alterazione dell'olfatto, del gusto, maggiore suscettibilità allo sviluppo di malattie infettive.
- Cardiomiopatia dilatativa: gradualmente tutte le camere del cuore si riempiono sempre di più. Il cuore è teso. Ciò avviene come reazione protettiva e compensatoria del corpo per mantenere un livello sufficiente di circolazione sanguigna. A poco a poco, il costante allungamento del cuore porta alla perdita della sua capacità di contrarsi normalmente. Clinicamente, ciò si esprime con disturbi del ritmo e della conduzione, sindrome edematosa, dolore al cuore, affaticamento e grave debolezza generale.
- stadio anemico- il sintomo principale di questa fase è una diminuzione della produzione di tutte le cellule del sangue, che si trasforma nei seguenti sintomi:
- Anemia da carenza di ferro aplastica - si sviluppa a seguito dell'inibizione dei processi emopoietici nel midollo osseo dovuta alla mielofibrosi - spostamento delle cellule ematopoietiche dal midollo osseo da parte del tessuto connettivo. Compaiono pallore della pelle, aumento dell'affaticamento, debolezza generale pronunciata, svenimento, sensazione di mancanza d'aria.
- Sanguinamento - si verifica con le più piccole lesioni sulla pelle e sulle mucose a causa della ridotta produzione di piastrine e della sintesi di piastrine che perdono le loro funzioni.
Importante! In assenza di trattamento, lo stadio terminale avviene molto rapidamente con lo sviluppo di un esito letale.
Eritremia nei bambini, caratteristiche
La vera policitemia nei neonati e nei bambini piccoli non è tipica. Se il bambino presenta sintomi della malattia, indicano lo sviluppo di un processo secondario che può verificarsi a causa di:- Ipossia.
- Dispepsia tossica.
- Feto - insufficienza placentare.
Importante! I gemelli hanno la policitemia congenita dovuta a difetti genetici, che si manifesta fin dalla nascita.
Fondamentalmente, la malattia si manifesta a 2 settimane di vita di un bambino.
La stadiazione della malattia nei bambini è del tutto identica a quella degli adulti, ma nei bambini la malattia è molto più grave, con lo sviluppo di gravi infezioni batteriche, difetti cardiaci, sclerosi del midollo osseo, che portano a morte prematura. Il trattamento per la policitemia è lo stesso degli adulti, come discusso di seguito.
Diagnosi della malattia di Wakez
Quando si diagnostica la policitemia, viene utilizzato un piano diagnostico ben definito, che comprende i seguenti passaggi: 
- Raccolta dati anamnestici.
- Ispezione visuale.
- Esame del sangue, che dovrebbe includere:
- Il numero di globuli rossi e di altre cellule del sangue.
- Ematocrito.
- Volume medio degli eritrociti - MCV.
- Il contenuto medio di emoglobina negli eritrociti - MCH.
- La concentrazione media di emoglobina negli eritrociti è MCHC.
- L'ampiezza di distribuzione degli eritrociti in volume è RDW.
- Eritropoietina nel siero del sangue.
- Test genetici molecolari del sangue per rilevare mutazioni.
- Esame ecografico degli organi addominali.
- Un esame del sangue biochimico, in particolare per l'acido urico, il cui aumento dei livelli indica lo sviluppo della gotta.
- Fibrogastroduodenoscopia.
- TAC addominale in modalità vascolare.
- Biopsia del midollo osseo.
- Valutazione delle funzioni della respirazione esterna.
- Determinazione del contenuto di ossigeno e anidride carbonica nel sangue.
- grandi arterie.
- EcoCG.
- Analisi generale delle urine.
Per fare una diagnosi di vera policitemia, dopo tutte le manipolazioni, vengono applicati alcuni criteri in base ai quali viene posta la diagnosi di policitemia:
- Grandi criteri:
- Livelli di emoglobina superiori a 185 g/l per gli uomini e 165 g/l per le donne, nonché altri segni di aumento della massa dei globuli rossi - ematocrito > 52% negli uomini, > 48% nelle donne.
- Rilevazione di mutazioni nel gene JAK2V617F.
- Piccoli criteri:
- La panmielosi in una biopsia del midollo osseo è un aumento della proliferazione di germogli ematopoietici eritroidi, granulocitici e megacariocitici.
- Bassi valori di eritropoietina.
- La formazione di colonie di eritrociti endogene senza la partecipazione dell'eritropoietina nello studio della biopsia delle colture di midollo osseo.
Importante! La diagnosi è pienamente confermata in presenza di due criteri maggiori e uno minore.
Trattamento
Il trattamento dei pazienti con policitemia avviene nelle condizioni del dipartimento di ematologia. Per il trattamento di questa condizione, vengono utilizzate le seguenti misure:- Salasso: eseguito per ridurre il numero di globuli rossi e di emoglobina. La procedura viene eseguita una volta ogni 1-2 giorni con la raccolta fino a 500 ml di sangue.
- La citoferesi è il passaggio del sangue attraverso filtri speciali, con l'aiuto dei quali vengono eliminati alcuni globuli rossi.
- Ricezione di citostatici: ciclofosano, ciclofosfamide, idrossiurea, ecc.
- Terapia antipiastrinica con aspirina, dipiridamolo.
- Interferoni.
- trattamento sintomatico.
Importante!È severamente vietato curare autonomamente la malattia senza intervento medico, nonché utilizzare metodi e tipi di trattamento dubbi.
Importante nel trattamento della policitemia è una dieta che escluda completamente l'assunzione di alimenti che aumentano la formazione del sangue. Con l'aggiunta della gotta, la carne ed il pesce in genere possono essere esclusi dalla dieta dei pazienti e sostituiti con alimenti vegetali.

In generale, la base del trattamento è la distinzione tra processo primario e secondario, poiché nella policitemia secondaria viene trattata principalmente la condizione che ha causato lo sviluppo dell'eritremia.
Complicazioni
La policitemia è caratterizzata da un'alta probabilità di complicazioni formidabili come:
- Ipertensione arteriosa in forma grave.
- Ictus emorragici.
- Infarto miocardico.
- Leucemia mieloide acuta.
- Leucemia mieloide cronica.
- Eritromielosi.
In alcuni casi, anche un trattamento tempestivo porta allo sviluppo di situazioni pericolose che possono finire con la morte in qualsiasi momento.
Previsione
La prognosi della policitemia dipende direttamente dal tipo, dal decorso, dalla tempestività e dalla correttezza del trattamento.
Importante! Senza un trattamento adeguato, circa il 50% dei pazienti muore entro un anno e mezzo dal momento della diagnosi.
Con una terapia tempestiva, la prognosi nei pazienti con eritremia è abbastanza favorevole e dimostra un tasso di sopravvivenza a 10 anni in oltre il 75% dei casi.
La policitemia vera (policitemia primaria, malattia di Wakez, eritremia) è la malattia più comune nel gruppo delle malattie mieloproliferative croniche. Il processo patologico colpisce principalmente il midollo osseo eritroblastico, che provoca un aumento del numero degli eritrociti nel sangue periferico, nonché un aumento della viscosità e della massa del sangue circolante (ipervolemia).
La malattia si manifesta prevalentemente negli anziani (l'età media di insorgenza è di circa 60 anni), ma viene diagnosticata anche nei giovani e nei bambini. Per i pazienti più giovani è caratteristico un decorso più grave della malattia. Gli uomini sono leggermente più inclini alla policitemia vera rispetto alle donne, ma i pazienti giovani sono caratterizzati da una proporzione inversa.
Cause e fattori di rischio
Le cause che contribuiscono alla comparsa della vera policitemia non sono state definitivamente stabilite. La patologia può essere sia ereditaria che acquisita. Riscontrata predisposizione familiare alla malattia. Nei pazienti con vera policitemia vengono rilevate mutazioni genetiche ereditate con modalità autosomica recessiva.
I fattori di rischio includono:
- impatto sul corpo di sostanze tossiche;
- Radiazione ionizzante;
- esposizione ai raggi X;
- ustioni estese;
- uso a lungo termine di numerosi farmaci (sali d'oro, ecc.);
- forme avanzate di tubercolosi;
- angoscia;
- malattie virali;
- neoplasie tumorali;
- fumare;
- disturbi endocrini causati da tumori delle ghiandole surrenali;
- difetti cardiaci;
- malattie del fegato e/o dei reni;
- interventi chirurgici estesi.
Forme della malattia
La policitemia vera è di due tipi:
- primario (non conseguenza di altre patologie);
- secondario (si sviluppa sullo sfondo di altre malattie).
Senza una terapia adeguata per la policitemia vera, il 50% dei pazienti muore entro 1-1,5 anni dal momento della diagnosi.
Fasi della malattia
Ci sono tre fasi nel quadro clinico della policitemia vera:
- Iniziale (malosintomatico): le manifestazioni cliniche sono insignificanti, la durata è di circa 5 anni.
- Lo stadio eritremico (esteso) della durata di 10-20 anni, a sua volta, è suddiviso in sottostadi: IIA - assenza di metaplasia mieloide della milza; IIB - presenza di metaplasia mieloide della milza;
- Stadio della metaplasia mieloide posteritremica (anemica) con o senza mielofibrosi; in grado di svilupparsi in leucemia cronica o acuta.
Sintomi
La policitemia vera è caratterizzata da un lungo decorso asintomatico. Il quadro clinico è associato ad un aumento della produzione di globuli rossi nel midollo osseo, che spesso è accompagnato da un aumento del numero di altri elementi cellulari nel sangue. Un aumento della conta piastrinica porta alla trombosi vascolare, che può causare ictus, infarto miocardico, attacchi ischemici transitori, ecc.

Nelle fasi successive della malattia possono verificarsi:
- prurito cutaneo, aggravato dall'esposizione all'acqua;
- attacchi di dolore pressante dietro lo sterno durante lo sforzo fisico;
- debolezza, aumento dell'affaticamento;
- disturbo della memoria;
- mal di testa, vertigini;
- eritrocianosi;
- arrossamento degli occhi;
- deficit visivo;
- aumento della pressione sanguigna;
- sanguinamento spontaneo, ecchimosi, sanguinamento gastrointestinale;
- vene varicose (soprattutto vene del collo);
- dolore intenso a breve termine alla punta delle dita;
- ulcera allo stomaco e/o ulcera duodenale;
- dolori articolari;
- insufficienza cardiaca.
Diagnostica
La diagnosi di policitemia vera viene stabilita sulla base dei dati ottenuti durante l'esame:
- raccolta dell'anamnesi;
- esame obiettivo;
- esame del sangue generale e biochimico;
- analisi generale delle urine;
- trepanobiopsia seguita dall'analisi istologica della biopsia;
- esame ecografico;
- imaging a risonanza magnetica o computerizzata;
- analisi genetica molecolare.
 Un esame del sangue per la policitemia vera mostra un aumento del numero di globuli rossi
Un esame del sangue per la policitemia vera mostra un aumento del numero di globuli rossi
Criteri diagnostici per la policitemia vera:
- aumento della massa degli eritrociti circolanti: negli uomini - più di 36 ml / kg, nelle donne - più di 32 ml / kg;
- leucociti - 12 × 10 9 / le più;
- piastrine - 400 × 10 9 / le più;
- aumento dell'emoglobina fino a 180–240 g/l;
- un aumento della saturazione di ossigeno nel sangue arterioso - 92% o più;
- un aumento del contenuto sierico di vitamina B 12 - 900 pg / ml o più;
- aumento dell'attività della fosfatasi alcalina dei leucociti fino a 100;
- splenomegalia.
La malattia si manifesta prevalentemente negli anziani (l'età media di insorgenza è di circa 60 anni), ma viene diagnosticata anche nei giovani e nei bambini.
La diagnosi differenziale è richiesta con eritrocitosi assoluta e relativa (falsa), neoplasie, trombosi della vena epatica.
Trattamento
Il trattamento della policitemia vera è mirato principalmente a prevenire lo sviluppo della leucemia, nonché alla prevenzione e/o alla terapia delle complicanze tromboemorragiche. La terapia sintomatica viene effettuata al fine di migliorare la qualità della vita del paziente.
Per ridurre la viscosità del sangue nella sindrome da iperviscosità, viene eseguito un ciclo di salasso (esfusione, salasso). Tuttavia, con una trombocitosi inizialmente elevata, il salasso può contribuire alla comparsa di complicanze trombotiche. La terapia mielosoppressiva è indicata per i pazienti che non tollerano il salasso, così come nell'infanzia e nell'adolescenza.

I preparati di interferone sono prescritti per un lungo corso (2-3 mesi) per ridurre la mieloproliferazione, la trombocitemia e anche per prevenire lo sviluppo di complicanze vascolari.
Con l'aiuto di metodi terapeutici hardware (eritrocitoaferesi, ecc.), Le cellule del sangue in eccesso vengono rimosse. Per prevenire la trombosi vengono prescritti anticoagulanti. Per ridurre le manifestazioni di prurito vengono utilizzati antistaminici. Inoltre, si consiglia ai pazienti di aderire a una dieta vegetariana a base di latte e di limitare l'attività fisica.
Con un pronunciato aumento delle dimensioni della milza (ipersplenismo), per i pazienti è indicata la splenectomia.
Possibili complicazioni e conseguenze
La policitemia vera può essere complicata da:
- mielofibrosi;
- infarto della milza;
- anemia;
- nefrosclerosi;
- colelitiasi e/o urolitiasi;
- gotta;
- infarto miocardico;
- ictus ischemico;
- cirrosi epatica;
- embolia polmonare;
- leucemia acuta o cronica.
Previsione
Con diagnosi e trattamento tempestivi, la sopravvivenza supera i 10 anni. Senza una terapia adeguata, il 50% dei pazienti muore entro 1-1,5 anni dal momento della diagnosi.
Prevenzione
Dato che le cause esatte della malattia non sono chiare, non sono ancora stati sviluppati metodi efficaci per la prevenzione della policitemia vera.
Video da YouTube sull'argomento dell'articolo:
- In contatto con 0
- Google+ 0
- OK 0
- Facebook 0






