
Programma delle lezioni 1. Prodotti finali del metabolismo dell'azoto: sali di ammonio, urea e acido urico. 1. Prodotti finali del metabolismo dell'azoto: sali di ammonio, urea e acido urico. 2. Neutralizzazione dell'ammoniaca: sintesi di glutammina e carbamil fosfato, amminazione riduttiva del 2-ossoglutarato. 2. Neutralizzazione dell'ammoniaca: sintesi di glutammina e carbamil fosfato, amminazione riduttiva del 2-ossoglutarato. 3. Glutammina come donatore di gruppi ammidici nella sintesi di numerosi composti. Rene glutaminasi, formazione ed escrezione di sali di ammonio. Attivazione adattativa della glutaminasi renale nell'acidosi. 3. Glutammina come donatore di gruppi ammidici nella sintesi di numerosi composti. Rene glutaminasi, formazione ed escrezione di sali di ammonio. Attivazione adattativa della glutaminasi renale nell'acidosi.

Piano di lezione 4. Biosintesi dell'urea. 4. Biosintesi dell'urea. 5. Collegamento del ciclo dell'ornitina con le trasformazioni degli acidi fumarico e aspartico; origine degli atomi di azoto ureico. 5. Collegamento del ciclo dell'ornitina con le trasformazioni degli acidi fumarico e aspartico; origine degli atomi di azoto ureico. 6. Biosintesi dell'urea come meccanismo per prevenire la formazione di ammoniaca. Uremia. 6. Biosintesi dell'urea come meccanismo per prevenire la formazione di ammoniaca. Uremia.

PRODOTTI FINALI: AMMONIACA PRODOTTI FINALI: AMMONIACA La degradazione degli amminoacidi avviene prevalentemente nel fegato. Questo rilascia ammoniaca direttamente o indirettamente. Quantità significative di ammoniaca si formano durante la scomposizione delle purine e delle piramidine. La degradazione degli amminoacidi avviene principalmente nel fegato. Questo rilascia ammoniaca direttamente o indirettamente. Quantità significative di ammoniaca si formano durante la scomposizione delle purine e delle piramidine.


TOSSICITÀ DELL'AMMONIACA L'ammoniaca - NH 3 è un veleno cellulare. Ad alte concentrazioni danneggia principalmente le cellule nervose (coma epatargico). Ammoniaca - NH 3 è un veleno cellulare. Ad alte concentrazioni danneggia principalmente le cellule nervose (coma epatargico). Normalmente, la scomposizione di 70 g di AA al giorno porta a una concentrazione di NH 3 nel sangue di 60 µmol/l, che è 100 volte inferiore alla concentrazione di glucosio nel sangue. Normalmente, la scomposizione di 70 g di AA al giorno porta a una concentrazione di NH 3 nel sangue di 60 µmol/l, che è 100 volte inferiore alla concentrazione di glucosio nel sangue.

Tossicità dell'ammoniaca Negli esperimenti sulla concentrazione nei conigli Negli esperimenti sui conigli, la concentrazione di NH 3 3 mmol/l ha provocato la morte! NH 3 3 mmol/l ha causato la morte! Cause di tossicità: Cause di tossicità: 1. a pH del sangue sotto forma di NH 4 +, penetra attraverso il plasma. e membrane MX con grande difficoltà. 1. al pH del sangue sotto forma di NH 4 +, penetra attraverso il plasma. e membrane MX con grande difficoltà.

Neutro. dicono gratuito NH 3 passa facilmente attraverso queste membrane. A pH 7,4, solo l'1% di NH3 della quantità totale di ammoniaca penetra nelle cellule cerebrali e nel MC. Neutro. dicono gratuito NH 3 passa facilmente attraverso queste membrane. A pH 7,4, solo l'1% di NH3 della quantità totale di ammoniaca penetra nelle cellule cerebrali e nel MC.

Cause di tossicità 2. NH 3 + a-KG + NADPH NH 3 + a-KG + NADPH 2 - Glu H 2 O Glu + NADP + H 2 O Deflusso di alpha-KG dal fondo CTC e conseguente diminuzione della velocità di ossidazione del glucosio

Tossicità dell'ammoniaca L'ammoniaca è così tossica che deve essere rimossa immediatamente mediante qualche meccanismo di escrezione o mediante incorporazione in qualche altro composto contenente azoto che non abbia una tossicità simile. L'ammoniaca è così tossica che deve essere rimossa immediatamente dall'uno o dall'altro meccanismo di escrezione, o per incorporazione in qualche altro composto contenente azoto che non abbia una tossicità simile.

Glu. 3. Amminazione a-KG --> Glu. 4. Amidazione delle proteine. 4. Amidir" title=" Meccanismi di disintossicazione da ammoniaca 1. Sintesi di glutammina: Gln, asparagina: Asn. 1. Sintesi di glutammina: Gln, asparagina: Asn. 2. Sintesi di urea. 2. Sintesi di urea 3. Aminazione di a-KG --> Glu 3. Aminazione di a-KG --> Glu 4. Amidazione di proteine 4. Amidir" class="link_thumb"> 11 !} Meccanismi di disintossicazione da ammoniaca 1. Sintesi di glutammina: Gln, asparagina: Asn. 1. Sintesi di glutammina: Gln, asparagina: Asn. 2. Sintesi dell'urea. 2. Sintesi dell'urea. 3. Amminazione a-KG --> Glu. 3. Amminazione a-KG --> Glu. 4. Amidazione delle proteine. 4. Amidazione delle proteine. Glu. 3. Amminazione a-KG --> Glu. 4. Amidazione delle proteine. 4. Amidir "> Glu. 3. Aminazione di a-KG --> Glu. 4. Amidazione delle proteine. 4. Amidazione delle proteine."> Glu. 3. Amminazione a-KG --> Glu. 4. Amidazione delle proteine. 4. Amidir" title=" Meccanismi di disintossicazione da ammoniaca 1. Sintesi di glutammina: Gln, asparagina: Asn. 1. Sintesi di glutammina: Gln, asparagina: Asn. 2. Sintesi di urea. 2. Sintesi di urea 3. Aminazione di a-KG --> Glu 3. Aminazione di a-KG --> Glu 4. Amidazione di proteine 4. Amidir"> title="Meccanismi di disintossicazione da ammoniaca 1. Sintesi di glutammina: Gln, asparagina: Asn. 1. Sintesi di glutammina: Gln, asparagina: Asn. 2. Sintesi dell'urea. 2. Sintesi dell'urea. 3. Amminazione a-KG --> Glu. 3. Amminazione a-KG --> Glu. 4. Amidazione delle proteine. 4. Amdir"> !}
Meccanismi di detoxication di ammoniaca 5. Sintesi di purin. e piramidi. strutture. 5. Sintesi delle purine. e piramidi. strutture. 6. Neutralizzazione nei reni con acidi ed escrezione di sali di ammonio nelle urine. 6. Neutralizzazione nei reni con acidi ed escrezione di sali di ammonio nelle urine.

Neutralizzazione dell'ammoniaca Negli organismi autotrofi, la maggior parte dell'ammoniaca risultante può essere riutilizzata per la sintesi di nuove strutture cellulari. Gli eterotrofi, d'altra parte, ricevono solitamente una quantità significativa di proteine \u200b\u200bcon il cibo, la cui assimilazione può facilmente portare all'accumulo di una grande quantità di prodotti finali del metabolismo dell'azoto. La rimozione di questi rifiuti richiede la realizzazione di un apposito apparato. Negli organismi autotrofi, la maggior parte dell'ammoniaca risultante può essere riutilizzata per la sintesi di nuove strutture cellulari. Gli eterotrofi, d'altra parte, ricevono solitamente una quantità significativa di proteine \u200b\u200bcon il cibo, la cui assimilazione può facilmente portare all'accumulo di una grande quantità di prodotti finali del metabolismo dell'azoto. La rimozione di questi rifiuti richiede la realizzazione di un apposito apparato.

Ammoniaca disintossicante Un organismo acquatico può espellere l'ammoniaca direttamente, poiché verrà immediatamente diluita con acqua, con effetti dannosi minimi o nulli sulle cellule. L'escrezione di ammoniaca da parte degli animali che vivono in zone aride richiederebbe l'uso delle proprie risorse idriche per allevarla. Un organismo che vive in un ambiente acquatico può espellere direttamente l'ammoniaca, poiché verrà immediatamente diluita con acqua, con effetti dannosi minimi o nulli sulle cellule. L'escrezione di ammoniaca da parte degli animali che vivono in zone aride richiederebbe l'uso delle proprie risorse idriche per allevarla. Pertanto, in molte specie, l'ammoniaca viene convertita nel corpo in altri composti meno tossici. Pertanto, in molte specie, l'ammoniaca viene convertita nel corpo in altri composti meno tossici.

Amminazione riduttiva La maggior parte degli organismi ha la capacità di riciclare l'ammoniaca attraverso una reazione catalizzata dalla glutammato deidrogenasi. La maggior parte degli organismi ha la capacità di riciclare l'ammoniaca attraverso una reazione catalizzata dalla glutammato deidrogenasi. A-chetoglutarato + NH3 + NADPH.H+ A-chetoglutarato + NH3 + NADPH.H+ Glutammato + NADP+. Glutammato + NADP +. Questa è amminazione riduttiva. Questa è amminazione riduttiva. Tuttavia, una parte dell'ammoniaca formata rimane inutilizzata e alla fine viene espulsa dal corpo di invertebrati e vertebrati in forma libera, o sotto forma di acido urico o sotto forma di urea. Tuttavia, una parte dell'ammoniaca formata rimane inutilizzata e alla fine viene espulsa dal corpo di invertebrati e vertebrati in forma libera, o sotto forma di acido urico o sotto forma di urea.







UREA UREA Nell'uomo l'inattivazione dell'ammoniaca è dovuta principalmente alla sintesi dell'urea, parte dell'NH 3 viene escreta direttamente dai reni. Nell'uomo, l'inattivazione dell'ammoniaca viene effettuata principalmente a causa della sintesi dell'urea, parte di NH 3 viene escreta direttamente dai reni.

ORGANISMI AMMONIOTELICI In diverse specie di vertebrati, l'ammoniaca viene inattivata ed escreta in modi diversi. Gli animali che vivono nell'acqua emettono ammoniaca direttamente nell'acqua; ad esempio, nei pesci viene espulso attraverso le branchie (organismi ammoniotelici). In diverse specie di vertebrati, l'ammoniaca viene inattivata ed escreta in modi diversi. Gli animali che vivono nell'acqua emettono ammoniaca direttamente nell'acqua; ad esempio, nei pesci viene espulso attraverso le branchie (organismi ammoniotelici).

ORGANISMI UREOTELICI I vertebrati terrestri, compreso l'uomo, espellono solo una piccola quantità di ammoniaca e la maggior parte viene convertita in urea (organismi ureoteliali). I vertebrati terrestri, compreso l'uomo, espellono solo una piccola quantità di ammoniaca e la maggior parte viene convertita in urea (organismi ureotelici).

ORGANISMI URICOTELICI Uccelli e rettili, al contrario, formano acido urico che, a causa della conservazione dell'acqua, viene escreto principalmente in forma solida (organismi uricotelici). Uccelli e rettili, al contrario, formano acido urico che, a causa della conservazione dell'acqua, viene escreto principalmente in forma solida (organismi uricotelici).

Sintesi dell'urea L'urea, a differenza dell'ammoniaca, è un composto neutro e non tossico. Una piccola molecola di urea può passare attraverso le membrane e, grazie alla sua buona solubilità in acqua, l'urea viene facilmente trasportata nel sangue ed escreta nelle urine. L'urea, a differenza dell'ammoniaca, è un composto neutro e non tossico. Una piccola molecola di urea può passare attraverso le membrane e, grazie alla sua buona solubilità in acqua, l'urea viene facilmente trasportata nel sangue ed escreta nelle urine.

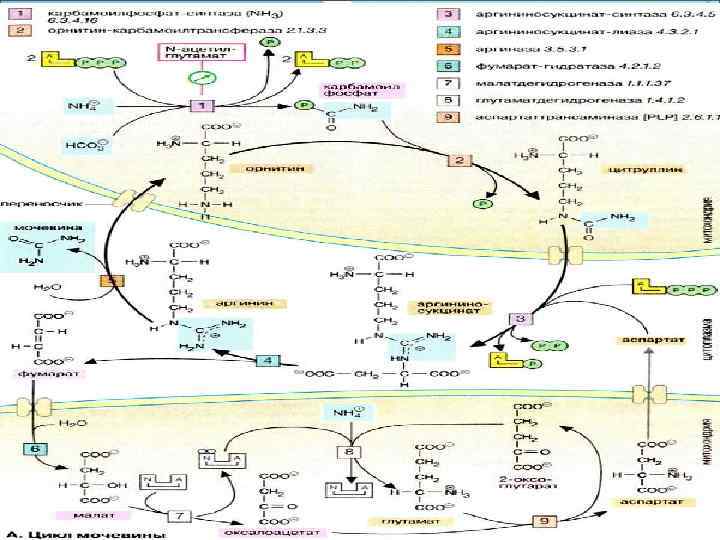
FASI DELLA SINTESI DELL'UREA L'urea si forma come risultato di una sequenza ciclica di reazioni che si verificano nel fegato. L'urea si forma come risultato di una sequenza ciclica di reazioni che si verificano nel fegato. Entrambi gli atomi di azoto sono prelevati dall'ammoniaca libera e attraverso la deaminazione dell'aspartato, il gruppo carbonilico dal bicarbonato. Entrambi gli atomi di azoto sono prelevati dall'ammoniaca libera e attraverso la deaminazione dell'aspartato, il gruppo carbonilico dal bicarbonato.

La prima reazione Nella prima fase, la reazione, il carbamil fosfato è formato da bicarbonato (HCO3-) e ammoniaca con il consumo di 2 molecole di ATP. Nella prima fase, la reazione, il carbamil fosfato è formato da bicarbonato (HCO3-) e ammoniaca con il consumo di 2 molecole di ATP.


Seconda fase Seconda fase Nella fase successiva, la reazione, il residuo carbamoilico viene trasferito all'ornitina per formare la citrullina. Questa reazione richiede nuovamente energia sotto forma di ATP, che viene poi scomposta in AMP e difosfato. Nella fase successiva, la reazione, il residuo carbamoilico viene trasferito all'ornitina per formare la citrullina. Questa reazione richiede nuovamente energia sotto forma di ATP, che viene poi scomposta in AMP e difosfato.











LA BICICLETTA DI KREBS Il fumarato formatosi nel ciclo dell'urea può, come risultato di due stadi del ciclo del citrato, passare attraverso il malato all'ossalacetato, che, a causa della transaminazione, viene ulteriormente terminato in aspartato. Quest'ultimo è anche coinvolto nuovamente nel ciclo dell'urea. Il fumarato formato nel ciclo dell'urea può, come risultato di due stadi del ciclo del citrato, passare attraverso il malato all'ossalacetato, che, a causa della transaminazione, è ulteriormente terminato in aspartato. Quest'ultimo è anche coinvolto nuovamente nel ciclo dell'urea.

PROCESSO DIPENDENTE DALL'ENERGIA La biosintesi dell'urea richiede molta energia. L'energia viene fornita dalla scissione di quattro legami ad alta energia: due nella sintesi del carbamil fosfato e due (!) nella formazione dell'argininosuccinato (ATP AMP + PPi, PPi 2Pi). La biosintesi dell'urea richiede molta energia. L'energia viene fornita dalla scissione di quattro legami ad alta energia: due nella sintesi del carbamil fosfato e due (!) nella formazione dell'argininosuccinato (ATP AMP + PPi, PPi 2Pi).

COMPARTMENTALIZZAZIONE Il ciclo dell'urea avviene esclusivamente nel fegato. È diviso in due compartimenti: mitocondri e citoplasma. Il passaggio attraverso la membrana di composti intermedi di citrullina e ornitina è possibile solo con l'ausilio di trasportatori. Il ciclo dell'urea si svolge esclusivamente nel fegato. È diviso in due compartimenti: mitocondri e citoplasma. Il passaggio attraverso la membrana di composti intermedi di citrullina e ornitina è possibile solo con l'ausilio di trasportatori.

REGOLAZIONE ALLOSTERICA DELLA SINTESI DELL'UREA La velocità di sintesi dell'urea è determinata dalla prima reazione del ciclo. La carbamoil fosfato sintasi è attiva solo in presenza di N-acetilglutammato. Lo stato metabolico (livelli di arginina, apporto energetico) dipende fortemente dalla concentrazione di questo effettore allosterico. La velocità di sintesi dell'urea è determinata dalla prima reazione del ciclo. La carbamoil fosfato sintasi è attiva solo in presenza di N-acetilglutammato. Lo stato metabolico (livelli di arginina, apporto energetico) dipende fortemente dalla concentrazione di questo effettore allosterico. La velocità di sintesi dell'urea viene determinata per prima La velocità di sintesi dell'urea viene determinata per prima
 Metabolismo dell'azoto - un insieme di trasformazioni chimiche di sostanze contenenti azoto nel corpo. A. o. comprende lo scambio di proteine semplici e complesse, acidi nucleici, i loro prodotti di decadimento (peptidi, amminoacidi e nucleotidi), sostanze grasse contenenti azoto (lipidi), amminozuccheri, ormoni, vitamine, ecc. Per il normale corso della vita processi, il corpo deve essere fornito della quantità necessaria di azoto digeribile. Il componente principale e la principale fonte di azoto nel cibo umano sono le sostanze proteiche.
Metabolismo dell'azoto - un insieme di trasformazioni chimiche di sostanze contenenti azoto nel corpo. A. o. comprende lo scambio di proteine semplici e complesse, acidi nucleici, i loro prodotti di decadimento (peptidi, amminoacidi e nucleotidi), sostanze grasse contenenti azoto (lipidi), amminozuccheri, ormoni, vitamine, ecc. Per il normale corso della vita processi, il corpo deve essere fornito della quantità necessaria di azoto digeribile. Il componente principale e la principale fonte di azoto nel cibo umano sono le sostanze proteiche.

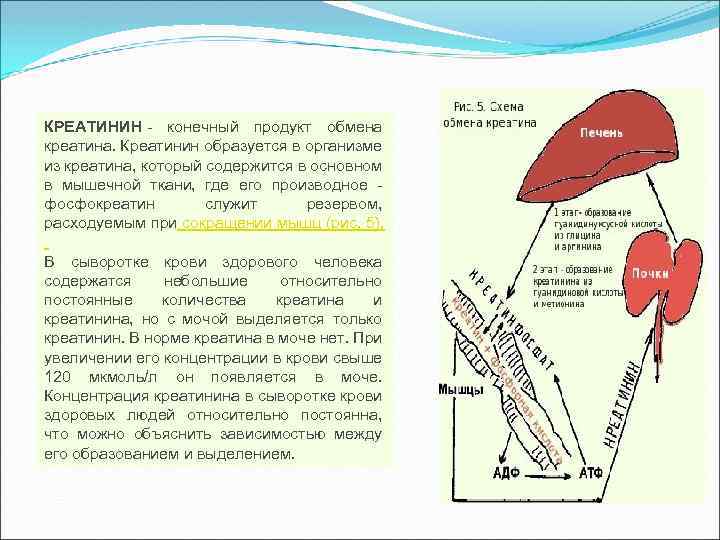 La creatinina è il prodotto finale del metabolismo della creatina. La creatinina si forma nell'organismo dalla creatina, che si trova principalmente nel tessuto muscolare, dove il suo derivato, la fosfocreatina, funge da riserva consumata durante la contrazione muscolare (Fig. 5). Il siero del sangue di una persona sana contiene quantità piccole e relativamente costanti di creatina e creatinina, ma solo la creatinina viene escreta nelle urine. Normalmente, non c'è creatina nelle urine. Con un aumento della sua concentrazione nel sangue superiore a 120 µmol / l, appare nelle urine. La concentrazione di creatinina nel siero del sangue di persone sane è relativamente costante, il che può essere spiegato dalla relazione tra la sua formazione e l'escrezione.
La creatinina è il prodotto finale del metabolismo della creatina. La creatinina si forma nell'organismo dalla creatina, che si trova principalmente nel tessuto muscolare, dove il suo derivato, la fosfocreatina, funge da riserva consumata durante la contrazione muscolare (Fig. 5). Il siero del sangue di una persona sana contiene quantità piccole e relativamente costanti di creatina e creatinina, ma solo la creatinina viene escreta nelle urine. Normalmente, non c'è creatina nelle urine. Con un aumento della sua concentrazione nel sangue superiore a 120 µmol / l, appare nelle urine. La concentrazione di creatinina nel siero del sangue di persone sane è relativamente costante, il che può essere spiegato dalla relazione tra la sua formazione e l'escrezione.
 L'urea è il prodotto finale della disgregazione proteica nel corpo, che, una volta espulso attraverso i reni, rimuove i resti di azoto "non necessario". Formato nel fegato. Viene escreto insieme all'urina e in parte al sudore (che conferisce al sudore un odore specifico). L'urea aiuta il corpo a trattenere l'acqua e alcuni oligoelementi. Questo viene fatto attraverso il riassorbimento da parte dei reni nel flusso sanguigno. Ogni volta che avviene il processo di assorbimento, l'urea "tira" indietro nel flusso sanguigno molecole d'acqua e minerali utili. Tuttavia, il suo contenuto eccessivo danneggia organi e tessuti.
L'urea è il prodotto finale della disgregazione proteica nel corpo, che, una volta espulso attraverso i reni, rimuove i resti di azoto "non necessario". Formato nel fegato. Viene escreto insieme all'urina e in parte al sudore (che conferisce al sudore un odore specifico). L'urea aiuta il corpo a trattenere l'acqua e alcuni oligoelementi. Questo viene fatto attraverso il riassorbimento da parte dei reni nel flusso sanguigno. Ogni volta che avviene il processo di assorbimento, l'urea "tira" indietro nel flusso sanguigno molecole d'acqua e minerali utili. Tuttavia, il suo contenuto eccessivo danneggia organi e tessuti.
 Acido urico Prodotto finale del metabolismo delle basi puriniche che costituiscono i nucleotidi. A causa dell'escrezione di acido urico, l'eccesso di azoto viene rimosso dal corpo. Nel plasma, l'acido urico si trova prevalentemente sotto forma di sale sodico. La concentrazione di acido urico nel sangue è dovuta all'equilibrio dei processi di sintesi dell'acido urico e alla sua escrezione da parte dei reni.
Acido urico Prodotto finale del metabolismo delle basi puriniche che costituiscono i nucleotidi. A causa dell'escrezione di acido urico, l'eccesso di azoto viene rimosso dal corpo. Nel plasma, l'acido urico si trova prevalentemente sotto forma di sale sodico. La concentrazione di acido urico nel sangue è dovuta all'equilibrio dei processi di sintesi dell'acido urico e alla sua escrezione da parte dei reni.
 MODI FORMAZIONE DELL'AMMONIACA acido glutammico -acido chetoglutarico + NH 3 -amminoacido -chetoacido + NH 3 cisteina piruvato + NH 3 istidina acido urocaico + NH 3 glicina acido gliossalico + NH 3 glucosamina-6 -fosfato glucosio-6 - fosfato + NH 3 glutammina acido glutammico + NH 3 O ║ NH 2 -C O-P + CO 2 + NH 3 carbomoil fosfato glutammina ATP ADP urea pirimidine acido folico purine glucosamina
MODI FORMAZIONE DELL'AMMONIACA acido glutammico -acido chetoglutarico + NH 3 -amminoacido -chetoacido + NH 3 cisteina piruvato + NH 3 istidina acido urocaico + NH 3 glicina acido gliossalico + NH 3 glucosamina-6 -fosfato glucosio-6 - fosfato + NH 3 glutammina acido glutammico + NH 3 O ║ NH 2 -C O-P + CO 2 + NH 3 carbomoil fosfato glutammina ATP ADP urea pirimidine acido folico purine glucosamina
Src="http://present5.com/presentation/229073585_437016682/image-8.jpg" alt="CAUSE DI INTOSSICAZIONE DA AMMONIACA IPERAMMONIEMIA (>25 -40 µmol/l) Violazione della funzione di formazione dell'urea di il fegato Violazione della funzione di escrezione di azoto"> ПРИЧИНЫ АММИАЧНОЙ ИНТОКСИКАЦИИ ГИПЕРАММОНИЙЕМИЯ (>25 -40 мкмоль/л) Нарушение мочевинообразующей функции печени Нарушение азотовыделительной функции почек Острая почечная недостаточность Вирусный гепатит Хроническая почечная недостаточность Токсический гепатит Цирроз печени Портакавальные анастомозы на фоне богатой белками пищи Врожденные гипераммонийемии!}
 Azoto residuo e suoi componenti Indicatore Contenuto nel siero del sangue in mg/100 ml Unità SI Azoto residuo 20-40 mg/100 ml 7,06-14,1 mmol/l Urea 20-40 mg/100 ml 3,3-6,6 mmol/l Aminoazoto acidi 2,0-4,3 mg/100 ml 1,43-3,07 mmol/l Acido urico 2-6,4 mg/100 ml 0,12-0,38 mmol/l uomini 0 , 2-0,7 mg/100 ml 13-53 µmol/l donne 0,4-0,9 mg /100 ml 27-71 µmol/l uomini 1-2 mg/100 ml 0,088-0,177 mmol /l donne 0,5-1,6 mg/100 ml 0,044-0,141 mmol/l Ammoniaca 0,03-0,06 mg/100 ml 21,4-42,8 Creatina: Creatinina: Altre sostanze non proteiche (polipeptidi, nucleotidi, ecc.) Reazione xantoproteica 20 unità. Creatina: sangue intero 3-4 mg % 229-305 µmol/l plasma 1- 1,5 mg% 76,3- 114,5 µmol/l Azoto ureico nel sangue (urea: 2, 14) 9-14 mg % 3, 18- 4,94 mmol/l
Azoto residuo e suoi componenti Indicatore Contenuto nel siero del sangue in mg/100 ml Unità SI Azoto residuo 20-40 mg/100 ml 7,06-14,1 mmol/l Urea 20-40 mg/100 ml 3,3-6,6 mmol/l Aminoazoto acidi 2,0-4,3 mg/100 ml 1,43-3,07 mmol/l Acido urico 2-6,4 mg/100 ml 0,12-0,38 mmol/l uomini 0 , 2-0,7 mg/100 ml 13-53 µmol/l donne 0,4-0,9 mg /100 ml 27-71 µmol/l uomini 1-2 mg/100 ml 0,088-0,177 mmol /l donne 0,5-1,6 mg/100 ml 0,044-0,141 mmol/l Ammoniaca 0,03-0,06 mg/100 ml 21,4-42,8 Creatina: Creatinina: Altre sostanze non proteiche (polipeptidi, nucleotidi, ecc.) Reazione xantoproteica 20 unità. Creatina: sangue intero 3-4 mg % 229-305 µmol/l plasma 1- 1,5 mg% 76,3- 114,5 µmol/l Azoto ureico nel sangue (urea: 2, 14) 9-14 mg % 3, 18- 4,94 mmol/l
 iperazotemia da produzione Insufficienza epatocellulare delle frazioni non ureiche dell'azoto residuo (ammonio, azoto ammonico, ammine biogeniche); ↓frazione di urea nell'azoto residuo Aumento del catabolismo proteico (fame, sovralimentazione) delle frazioni non ureiche dell'azoto residuo (ammonio, azoto ammonico, ammine biogeniche); frazione urea nella ritenzione di azoto residuo Insufficienza renale acuta e cronica AKI: concentrazione plasmatica di urea, ↓clearance di urea, azoto residuo e azoto ureico CRF: azoto residuo fino a 200-300 mg/100 ml, tu, peptidi ("molecole medie") nel plasma
iperazotemia da produzione Insufficienza epatocellulare delle frazioni non ureiche dell'azoto residuo (ammonio, azoto ammonico, ammine biogeniche); ↓frazione di urea nell'azoto residuo Aumento del catabolismo proteico (fame, sovralimentazione) delle frazioni non ureiche dell'azoto residuo (ammonio, azoto ammonico, ammine biogeniche); frazione urea nella ritenzione di azoto residuo Insufficienza renale acuta e cronica AKI: concentrazione plasmatica di urea, ↓clearance di urea, azoto residuo e azoto ureico CRF: azoto residuo fino a 200-300 mg/100 ml, tu, peptidi ("molecole medie") nel plasma
 FONTI E METODI PER LA NEUTRALIZZAZIONE DELL'AMMONIACA NEI VARI TESSUTI Ammine biogeniche Aminoacidi Nucleotidi AMMONIACA Sintesi Formazione di urea glutammina alanina glutammato sali di ammonio (25 g/die) (0,5 g/die) fegato cervello muscoli, intestino cervello reni
FONTI E METODI PER LA NEUTRALIZZAZIONE DELL'AMMONIACA NEI VARI TESSUTI Ammine biogeniche Aminoacidi Nucleotidi AMMONIACA Sintesi Formazione di urea glutammina alanina glutammato sali di ammonio (25 g/die) (0,5 g/die) fegato cervello muscoli, intestino cervello reni
 REAZIONI DI LEGAME DELL'AMMONIACA NELLA CELLULA 1. La reazione di amminazione riduttiva di -chetoglutarato a L-glutammato: NADPH 2 NADPH NH 3 + -chetoglutarico glutammico + H 2 O acido glutammato deidrogenasi acido 2. La reazione della formazione di glutammina dall'acido glutammico con la partecipazione dell'enzima glutammina sintetasi. La reazione procede nel citosol delle cellule di tutti i tessuti, ma in misura maggiore nel cervello: COOH CONH 2 │ │ CH 2 ATP ADP + Fn CH 2 │ │ CH 2 2 CH │ │ HC-NH 2 glutammina sintetasi HC- NH 2 │ │ COOH COOH glutammina glutammina acido
REAZIONI DI LEGAME DELL'AMMONIACA NELLA CELLULA 1. La reazione di amminazione riduttiva di -chetoglutarato a L-glutammato: NADPH 2 NADPH NH 3 + -chetoglutarico glutammico + H 2 O acido glutammato deidrogenasi acido 2. La reazione della formazione di glutammina dall'acido glutammico con la partecipazione dell'enzima glutammina sintetasi. La reazione procede nel citosol delle cellule di tutti i tessuti, ma in misura maggiore nel cervello: COOH CONH 2 │ │ CH 2 ATP ADP + Fn CH 2 │ │ CH 2 2 CH │ │ HC-NH 2 glutammina sintetasi HC- NH 2 │ │ COOH COOH glutammina glutammina acido
 Vie di scambio di amminoacidi azotati e ammoniaca TESSUTO SANGUE FEGATO Muscoli, intestino Amino-KG Ala-KG NH 3 Ketok-you glutammato piruvato piruvato glutammato CO 2 AMP NH 3 glucosio carbomoil fosfato IMF glutammato glutammina CERVELLO e altri tessuti Amino-KG NH 3 Ketok-you glutammato glutammina -KG RENE NH 3 glutammato NH 3 URINE ciclo dell'ornitina NH 3 glutammato urea sali di ammonio glutammato urea
Vie di scambio di amminoacidi azotati e ammoniaca TESSUTO SANGUE FEGATO Muscoli, intestino Amino-KG Ala-KG NH 3 Ketok-you glutammato piruvato piruvato glutammato CO 2 AMP NH 3 glucosio carbomoil fosfato IMF glutammato glutammina CERVELLO e altri tessuti Amino-KG NH 3 Ketok-you glutammato glutammina -KG RENE NH 3 glutammato NH 3 URINE ciclo dell'ornitina NH 3 glutammato urea sali di ammonio glutammato urea
 CICLO DELL'UREA E SUA RELAZIONE CON IL TCA citoplasma NH 3 + CO 2 + H 2 O L-aspartato 2 ATP carbomoil fosfato citrullina argininosuccinato ossalacetato mitocondrio malato mitocondri ornitina arginina fumarato urea TCA
CICLO DELL'UREA E SUA RELAZIONE CON IL TCA citoplasma NH 3 + CO 2 + H 2 O L-aspartato 2 ATP carbomoil fosfato citrullina argininosuccinato ossalacetato mitocondrio malato mitocondri ornitina arginina fumarato urea TCA
IO. Lo scopo dello studio: Sapere prodotti finali del metabolismo proteico nel corpo, le principali fonti di formazione di ammoniaca, modi della sua neutralizzazione dal corpo.
II. Essere in grado di determinare quantitativamente il contenuto di urea mediante reazione cromatica con diacetil monoossima nel siero del sangue; conoscere le proprietà fisico-chimiche dell'urea.
III. Livello iniziale di conoscenza: reazioni qualitative all'ammoniaca (chimica inorganica).
IV. Risposta alle domande dei biglietti finali di controllo sul tema: “Decomposizione di proteine semplici. Metabolismo degli aminoacidi, prodotti finali del metabolismo dell'azoto.
1. I prodotti finali della decomposizione delle sostanze contenenti azoto sono anidride carbonica, acqua e ammoniaca, a differenza di carboidrati e lipidi. La fonte di ammoniaca nel corpo sono aminoacidi, basi azotate, ammine. L'ammoniaca si forma a seguito della deaminazione diretta e indiretta degli amminoacidi, (la fonte principale) della deaminazione idrolitica delle basi azotate, dell'inattivazione delle ammine biogeniche.
2. L'ammoniaca è tossica e la sua azione si manifesta in diversi sistemi funzionali: a) penetrando facilmente attraverso le membrane (violando il trasferimento transmembrana di Na + e K +) nei mitocondri, si lega all'α-chetoglutarato e ad altri chetoacidi (CTK), formare amminoacidi; in questi processi vengono utilizzati anche equivalenti riducenti (NADH+H+).
b) ad alte concentrazioni di ammoniaca, glutammato e aspartato formano ammidi, utilizzando ATP e distruggendo lo stesso TCA, che è la principale fonte di energia del cervello. c) L'accumulo di glutammato nel cervello aumenta la pressione osmotica, che porta allo sviluppo di edema. d) Un aumento della concentrazione di ammoniaca nel sangue (N - 0,4 - 0,7 mg / l) sposta il pH verso il lato alcalino, aumentando l'affinità dell'O 2 per l'emoglobina, che provoca ipossia del tessuto nervoso. e) Una diminuzione della concentrazione di α-chetoglutarato provoca l'inibizione del metabolismo degli aminoacidi (sintesi dei neurotrasmettitori), l'accelerazione della sintesi dell'ossalacetato dal piruvato, che è associata ad un aumento dell'uso di CO 2 .
3. L'iperammoniemia influisce principalmente negativamente sul cervello ed è accompagnata da nausea, vertigini, perdita di coscienza, ritardo mentale (in forma cronica).
4. La principale reazione di legame dell'ammoniaca in tutte le cellule è la sintesi della glutammina sotto l'azione della glutammina sintetasi nei mitocondri, dove l'ATP viene utilizzato per questo scopo. La glutammina entra nel flusso sanguigno per diffusione facilitata e viene trasportata nell'intestino e nei reni. Nell'intestino, sotto l'azione della glutaminasi, si forma il glutammato, che viene transaminato con il piruvato, trasformandolo in alanina, che viene assorbita dal fegato; Il 5% di ammoniaca viene rimosso attraverso l'intestino, il restante 90% viene escreto dai reni.
5. Nei reni, la glutammina viene anche idrolizzata con la formazione di ammoniaca sotto l'azione della glutaminasi, che viene attivata dall'acidosi. Nel lume dei tubuli, l'ammoniaca neutralizza i prodotti metabolici acidi, formando sali di ammonio per l'escrezione, riducendo la perdita di K + e Na +. (N - 0,5 g di sali di ammonio al giorno).
6. Un alto livello di glutammina nel sangue provoca il suo utilizzo in molte reazioni anaboliche come donatore di azoto (sintesi di basi azotate, ecc.)
7. Le quantità più significative di ammoniaca vengono neutralizzate nel fegato dalla sintesi di urea (86% di azoto nelle urine) in una quantità di ~25 g/die. La biosintesi dell'urea è un processo ciclico, in cui si trova la sostanza chiave ornitina, aggiungendo carbomoile formato da NH 3 e CO 2 dopo l'attivazione di 2ATP. La citrullina formata nei mitocondri viene trasportata al citosol per l'introduzione del secondo atomo di azoto dall'aspartato con la formazione di arginina. L'arginina viene idrolizzata dall'arginasi e riconvertita in ornitina, e il secondo prodotto di idrolisi è l'urea, che infatti in questo ciclo era formata da due atomi di azoto (fonti -NH 3 e aspartato) e un atomo di carbonio (da CO 2). L'energia è fornita da 3ATP (2 nella formazione di carbomol fosfato e 1 nella formazione di argininosuccinato).
8. Il ciclo dell'ornitina è strettamente correlato al ciclo TCA, da allora l'aspartato si forma durante la transaminazione del PAA dal TCA e il fumarato che rimane dall'aspartato dopo la rimozione di NH 3 ritorna al TCA e, quando viene convertito in PAA, si formano 3 ATP, che assicurano la biosintesi della molecola di urea .
9. I disturbi ereditari del ciclo dell'ornitina (citrullinemia, argininosuccinaturia, iperargininemia) portano all'iperamminemia e, nei casi più gravi, al coma epatico.
10. Il tasso di urea nel sangue è di 2,5-8,3 mmol / l. Una diminuzione si osserva nelle malattie del fegato, un aumento è il risultato di insufficienza renale.
Lavoro di laboratorio
metabolismo dell'azoto- un insieme di trasformazioni chimiche, reazioni di sintesi e decomposizione di composti azotati nel corpo; componente del metabolismo e dell'energia. Il concetto di "metabolismo dell'azoto" comprende il metabolismo proteico (un insieme di trasformazioni chimiche nel corpo delle proteine e dei loro prodotti metabolici), nonché lo scambio di peptidi, aminoacidi, acidi nucleici, nucleotidi, basi azotate, amminozuccheri (cfr. carboidrati), contenente azoto lipidi, vitamine, ormoni e altri composti contenenti azoto.
L'organismo degli animali e dell'uomo riceve azoto digeribile dal cibo, in cui la principale fonte di composti azotati sono le proteine di origine animale e vegetale. Il fattore principale per mantenere l'equilibrio dell'azoto - lo stato di AA, in cui la quantità di azoto in entrata e in uscita è la stessa - è un'adeguata assunzione di proteine dal cibo. In URSS, la norma giornaliera delle proteine nella dieta di un adulto è pari a 100 G, o 16 G azoto proteico, con un dispendio energetico di 2500 kcal. Il bilancio azotato (la differenza tra la quantità di azoto che entra nel corpo con il cibo e la quantità di azoto espulso dal corpo con urina, feci e sudore) è un indicatore dell'intensità di A. o. nell'organismo. La fame o un'alimentazione azotata insufficiente portano a un bilancio azotato negativo, o carenza di azoto, in cui la quantità di azoto escreta dal corpo supera la quantità di azoto che entra nel corpo con il cibo. Un bilancio azotato positivo, in cui la quantità di azoto introdotta con il cibo supera la quantità di azoto escreto dal corpo, si osserva durante il periodo di crescita corporea, durante i processi di rigenerazione dei tessuti, ecc. La condizione di A. circa. dipende in gran parte dalla qualità delle proteine alimentari, che a sua volta è determinata dalla sua composizione aminoacidica e, soprattutto, dalla presenza di aminoacidi essenziali.
È generalmente accettato che negli esseri umani e nei vertebrati A. o. inizia con la digestione dei composti azotati del cibo nel tratto gastrointestinale. Nello stomaco, le proteine vengono scomposte con la partecipazione di enzimi proteolitici digestivi. tripsina e gastrixin (cfr Proteolisi ) con la formazione di eptidi, oligopeptidi e singoli amminoacidi. Dallo stomaco, la massa alimentare entra nel duodeno e nelle sezioni sottostanti dell'intestino tenue, dove i peptidi subiscono un'ulteriore scissione catalizzata dagli enzimi del succo pancreatico tripsina, chimotripsina e carbossipeptidasi e dagli enzimi del succo intestinale aminopeptidasi e dipeptidasi. Enzimi). Insieme ai peptidi. l'intestino tenue scompone le proteine complesse (p. es., le nucleoproteine) e gli acidi nucleici. Anche la microflora intestinale contribuisce in modo significativo alla scomposizione dei biopolimeri contenenti azoto. Oligopeptidi, amminoacidi, nucleotidi, nucleosidi, ecc. Vengono assorbiti nell'intestino tenue, entrano nel sangue e vengono trasportati con esso in tutto il corpo. Anche le proteine dei tessuti corporei nel processo di costante rinnovamento subiscono la proteolisi sotto l'azione delle protses tissutali (peptidasi e catepsine) e i prodotti di degradazione delle proteine tissutali entrano nel sangue. Gli amminoacidi possono essere utilizzati per la nuova sintesi di proteine e altri composti (basi puriniche e pirimidiniche, nucleotidi, porfirine, ecc.), per produrre energia (ad esempio, attraverso l'inclusione nel ciclo degli acidi tricarbossilici) o possono essere sottoposti a ulteriore degradazione con la formazione di prodotti finali A. O., soggetti a escrezione dal corpo.
Gli aminoacidi che fanno parte delle proteine alimentari sono utilizzati per la sintesi delle proteine degli organi e dei tessuti del corpo. Sono inoltre coinvolti nella formazione di molti altri importanti composti biologici: nucleotidi purinici (glutammina, glicina, acido aspartico) e nucleotidi pirimidinici (glutammina, acido aspartico), serotonina (triptofano), melanina (fenilalpnina, tirosina), istamina (istidina) , adrenalina, noradrenalina, tiramina (tirosina), poliammine (arginina, metionina), colina (metionina), porfirine (glicina), creatina (glicina, arginina, metionina), coenzimi, zuccheri e polisaccaridi, lipidi, ecc. La reazione chimica più importante per l'organismo, alla quale partecipano quasi tutti gli amminoacidi, è la transaminazione, che consiste nel trasferimento enzimatico reversibile del gruppo a-ammino degli amminoacidi all'atomo di carbonio a dei chetoacidi o delle aldeidi. La transaminazione è una reazione fondamentale nella biosintesi degli amminoacidi non essenziali nel corpo. L'attività degli enzimi che catalizzano le reazioni di transaminazione è aminotransferasi - ha un grande valore clinico e diagnostico.
La degradazione degli amminoacidi può procedere attraverso diversi percorsi. La maggior parte degli amminoacidi può subire la decarbossilazione con la partecipazione degli enzimi decarbossilasi per formare ammine primarie, che possono quindi essere ossidate in reazioni catalizzate dalla monoaminossidasi o dalla diammina ossidasi. Quando le ammine biogeniche (istamina, serotonina, tiramina, acido g-amminobutirrico) vengono ossidate dalle ossidasi, si formano le aldeidi, che subiscono ulteriori trasformazioni, e ammoniaca, la principale via di ulteriore metabolismo è la formazione di urea.
Un'altra via principale per la degradazione degli amminoacidi è la deaminazione ossidativa con la formazione di ammoniaca e chetoacidi. La deaminazione diretta degli L-amminoacidi negli animali e nell'uomo procede molto lentamente, ad eccezione dell'acido glutammico, che viene intensamente deaminato con la partecipazione dell'enzima specifico glutammato deidrogenasi. La transaminazione preliminare di quasi tutti gli a-amminoacidi e l'ulteriore deaminazione dell'acido glutammico formatosi in acido a-chetoglutarico e ammoniaca è il meccanismo principale per la deaminazione degli amminoacidi naturali.
L'ammoniaca è il prodotto di vari percorsi di degradazione degli amminoacidi, che possono anche formarsi a seguito del metabolismo di altri composti contenenti azoto (ad esempio, durante la deaminazione dell'adenina, che fa parte della nicotinammide adenina dinucleotide - NAD). Il modo principale di legare e neutralizzare l'ammoniaca tossica negli animali ureotelici (animali in cui il prodotto finale di A. o è l'urea) è il cosiddetto ciclo dell'urea (sinonimo: ciclo dell'ornitina, ciclo di Krebs-Henseleit), che si verifica nel fegato . È una sequenza ciclica di reazioni enzimatiche, a seguito della quale l'urea viene sintetizzata dalla molecola di ammoniaca o dall'azoto ammidico della glutammina, dal gruppo amminico dell'acido aspartico e dall'anidride carbonica. Con un'assunzione giornaliera di 100 G l'escrezione proteica giornaliera di urea dal corpo è di circa 30 G. Nell'uomo e negli animali superiori esiste un altro modo per neutralizzare l'ammoniaca: la sintesi delle ammidi degli acidi dicarbossilici asparagan e glutammina dai corrispondenti amminoacidi. Negli animali uricotelici (rettili, uccelli), il prodotto finale di A. o. è acido urico.
Come risultato della rottura degli acidi nucleici e delle nucleoproteine nel tratto gastrointestinale, si formano nucleotidi e nucleosidi. Oligo- e mono-nucleotidi con la partecipazione di vari enzimi (esterasi, nucleotidasi, nucleosidasi, fosforilasi) vengono quindi convertiti in basi puriniche e pirimidiniche libere.
L'ulteriore percorso di degradazione delle basi puriniche di adenina e guanina consiste nella loro deaminazione idrolitica sotto l'influenza degli enzimi adenasi e guanasi con la formazione rispettivamente di ipoxantina (6-idrossipurina) e xantina (2,6-dioxipurina), che vengono poi convertiti in acido urico in reazioni catalizzate dalla xantina ossidasi. L'acido urico è uno dei prodotti finali di A. o. e il prodotto finale del metabolismo delle purine nell'uomo - viene escreto dal corpo con l'urina. La maggior parte dei mammiferi possiede l'enzima uricase, che catalizza la conversione dell'acido urico in allantoina escreta.
La degradazione delle basi pirimidiniche (uracile, timina) consiste nella loro riduzione con formazione di diidroderivati e successiva idrolisi, a seguito della quale si forma acido b-ureidopropionico dall'uracile e da ammoniaca, anidride carbonica e b-alanina. esso, e l'acido b-amminoisobutirrico è formato da timina, acido, anidride carbonica e ammoniaca. L'anidride carbonica e l'ammoniaca possono essere ulteriormente incluse nell'urea attraverso il ciclo dell'urea e la b-alanina è coinvolta nella sintesi dei più importanti composti biologicamente attivi: i dipeptidi contenenti istidina carnosina (b-alanil-L-istidina) e anserina (b -alanil-N-metil-L-istidina), presente nelle sostanze estrattive dei muscoli scheletrici, nonché nella sintesi dell'acido pantotenico e del coenzima A.
Pertanto, varie trasformazioni dei più importanti composti azotati del corpo sono interconnesse in un unico scambio. Processo complicato A. o. regolato a livello molecolare, cellulare e tissutale. Il regolamento di A. circa. in tutto l'organismo ha lo scopo di adattare l'intensità di A. o. alle mutevoli condizioni dell'ambiente e dell'ambiente interno ed è svolto dal sistema nervoso sia direttamente che agendo sulle ghiandole endocrine.
Negli adulti sani, il contenuto di composti azotati in organi, tessuti e fluidi biologici è a un livello relativamente costante. L'eccesso di azoto dal cibo viene escreto nelle urine e nelle feci e, con una mancanza di azoto nel cibo, il fabbisogno del corpo può essere coperto dall'uso di composti azotati nei tessuti corporei. Allo stesso tempo, la composizione urina cambia a seconda delle caratteristiche E. e bilancio azotato. Normalmente, con una dieta invariata e condizioni ambientali relativamente stabili, una quantità costante di prodotti finali di AA viene escreta dal corpo e lo sviluppo di condizioni patologiche porta al suo brusco cambiamento. Cambiamenti significativi nell'escrezione di composti azotati nelle urine, principalmente nell'escrezione di urea, possono essere osservati anche in assenza di patologia in caso di un cambiamento significativo nella dieta (ad esempio, quando la quantità di proteine consumate viene modificata ), e la concentrazione di azoto residuo (cfr. Azoto residuo ) nel sangue cambia leggermente.
In una ricerca E. è necessario tener conto della composizione quantitativa e qualitativa del cibo assunto, della composizione quantitativa e qualitativa dei composti azotati escreti nelle urine e nelle feci e contenuti nel sangue. Per la ricerca di A. su. utilizzare sostanze azotate etichettate con radionuclidi di azoto, fosforo, carbonio, zolfo, idrogeno, ossigeno e osservare la migrazione dell'etichetta e la sua incorporazione nella composizione dei prodotti finali di A. o. Gli amminoacidi marcati sono ampiamente utilizzati, ad esempio 15 N-glicina, che vengono introdotti nel corpo con il cibo o direttamente nel sangue. Una parte significativa dell'azoto glicina alimentare etichettato viene escreta come urea con l'urina e l'altra parte dell'etichetta entra nelle proteine dei tessuti ed è espulsa dal corpo molto lentamente. Condurre ricerche A. o. necessario per la diagnosi di molte condizioni patologiche e il monitoraggio dell'efficacia del trattamento, nonché lo sviluppo di diete razionali, incl. medicinale (cfr Nutrizione medica ).
Patologia A. o. (fino a molto significativo) provoca proteine. Può essere causato da malnutrizione generale, carenza prolungata di proteine o aminoacidi essenziali nella dieta, mancanza di carboidrati e grassi che forniscono energia per i processi di biosintesi proteica nel corpo. Le proteine possono essere dovute alla predominanza dei processi di degradazione proteica sulla loro sintesi, non solo come risultato di carenza alimentare di proteine e altri nutrienti essenziali, ma anche durante un lavoro muscolare pesante, lesioni, processi infiammatori e distrofici, ischemia, infezione, ah esteso , un difetto nella funzione trofica del sistema nervoso , insufficienza di ormoni anabolici (ormone della crescita, ormoni sessuali, insulina), sintesi eccessiva o assunzione eccessiva di ormoni steroidei dall'esterno, ecc. Violazione dell'assorbimento proteico nella patologia del tratto gastrointestinale (evacuazione accelerata del cibo dallo stomaco, condizioni ipo e anacide, blocco del dotto escretore del pancreas, indebolimento della funzione secretoria e aumento della motilità dell'intestino tenue nell'enterite e enterocolite, assorbimento alterato nell'intestino tenue, ecc.) può anche portare a carenza di proteine. Le proteine portano alla discordanza A. o. ed è caratterizzato da un pronunciato bilancio azotato negativo.
Sono noti casi di violazione della sintesi di alcune proteine (vedi. Immunopatologia, Fermentopatie), così come la sintesi geneticamente determinata di proteine anormali, ad esempio con emoglobinopatie, mieloma multiplo (cfr Emoblastosi paraproteinemiche ) e così via.
La patologia di A. o., che consiste in una violazione del metabolismo degli aminoacidi, è spesso associata ad anomalie nel processo di transaminazione: una diminuzione dell'attività delle aminotransferasi durante l'ipo- o l'avitaminosi B 6, una violazione della sintesi di questi enzimi, mancanza di chetoacidi per la transaminazione dovuta all'inibizione del ciclo dell'acido tricarbossilico durante l'ipossia e zucchero e, ecc. Una diminuzione dell'intensità della transaminazione porta all'inibizione della deaminazione dell'acido glutammico e questo, a sua volta, ad un aumento della proporzione di azoto amminoacidico nella composizione dell'azoto residuo nel sangue (iperaminoacidemia), iperazotemia generale e aminoaciduria. Iperaminoacidemia, aminoaciduria e azotemia generale sono caratteristiche di molti tipi di patologia di A.. Con estesi danni al fegato e altre condizioni associate a una massiccia disgregazione proteica nel corpo, i processi di deaminazione degli amminoacidi e la formazione di urea vengono interrotti in modo tale che la concentrazione di azoto residuo e il contenuto di azoto amminoacidico in esso aumentano sullo sfondo di una diminuzione del contenuto relativo di azoto ureico nell'azoto residuo (la cosiddetta produzione di azotemia).
L'azotemia di produzione è solitamente accompagnata dall'escrezione di aminoacidi in eccesso nelle urine, poiché anche in caso di normale funzionamento dei reni, la filtrazione degli aminoacidi nei glomeruli renali è più intensa del loro riassorbimento nei tubuli. Malattie renali, ostruzione delle vie urinarie, alterata circolazione renale portano allo sviluppo di azotemia da ritenzione, accompagnata da un aumento della concentrazione di azoto residuo nel sangue dovuto ad un aumento del contenuto di urea nel sangue (vedi. insufficienza renale ). Ferite estese, gravi e, infezioni, danni alle ossa tubolari, al midollo spinale e al cervello, la malattia di Itsenko-Cushing e molte altre gravi malattie sono accompagnate da aminoaciduria. È anche caratteristico delle condizioni patologiche che si verificano con processi di riassorbimento alterati nei tubuli renali: malattia di Wilson-Konovalov (vedi. Distrofia epatocerebrale ), Nefronoftisi Fanconi (cfr. Malattie simili al rachitismo ) e altri Queste malattie sono tra i numerosi disturbi geneticamente determinati di A. o. La violazione selettiva del riassorbimento della cistina e della cistinuria con un disturbo generalizzato del metabolismo della cistina sullo sfondo dell'aminoaciduria generale accompagna la cosiddetta cistinosi. In questa malattia, i cristalli di cistina si depositano nelle cellule del sistema reticoloendoteliale. malattia ereditaria fenilchetonuria caratterizzato da una violazione della conversione della fenilalanina in tirosina a seguito di una deficienza geneticamente determinata dell'enzima fenilalanina - 4-idrossilasi, che provoca l'accumulo nel sangue e nelle urine di fenilalanina non convertita e dei suoi prodotti metabolici - acido fenilpiruvico e fenilacetico. La violazione delle trasformazioni di questi composti è anche caratteristica dell'epatite virale.Tirosinemia, tirosinuria e tironosi si notano in ah, malattie diffuse del tessuto connettivo (collagenosi) e altre condizioni patologiche. Si sviluppano a causa della compromissione della transaminazione della tirosina. Un'anomalia congenita delle trasformazioni ossidative della tirosina è alla base dell'alcaptonuria, in cui un metabolita non convertito di questo amminoacido, l'acido omogentisico, si accumula nelle urine. Disturbi del metabolismo dei pigmenti nell'ipocorticismo (vedi. ghiandole surrenali ) sono associati all'inibizione della conversione della tirosina in melanina dovuta all'inibizione dell'enzima tirosinasi (la completa perdita della sintesi di questo pigmento è caratteristica di un'anomalia congenita della pigmentazione - a).
Con una massiccia rottura delle strutture cellulari (fame, lavoro muscolare pesante, infezioni, ecc.), Si nota un aumento patologico della concentrazione di azoto residuo dovuto ad un aumento del contenuto relativo di azoto dell'acido urico in esso (normalmente, la concentrazione di acido urico nel sangue non supera - 0,4 mmol/l).
Nella vecchiaia, l'intensità e il volume della sintesi proteica diminuiscono a causa dell'inibizione diretta della funzione biosintetica dell'organismo e dell'indebolimento della sua capacità di assorbire gli aminoacidi alimentari; si sviluppa un bilancio azotato negativo. I disturbi del metabolismo delle purine negli anziani portano all'accumulo e alla deposizione di sali di acido urico - urati nei muscoli, nelle articolazioni e nella cartilagine. Correzione di disturbi E. in età avanzata può essere effettuata attraverso diete speciali contenenti proteine animali di alta qualità, vitamine e oligoelementi, con un contenuto limitato di purine.
Il metabolismo dell'azoto nei bambini si distingue per una serie di caratteristiche, in particolare un bilancio azotato positivo come condizione necessaria per la crescita. L'intensità dei processi di A. o. durante la crescita del bambino subisce cambiamenti, particolarmente pronunciati nei neonati e nei bambini piccoli. Durante i primi 3 giorni di vita, il bilancio azotato è negativo, il che si spiega con un apporto insufficiente di proteine dal cibo. Durante questo periodo viene rilevato un aumento transitorio della concentrazione di azoto residuo nel sangue (la cosiddetta azotemia fisiologica), che talvolta raggiunge i 70 mmol/l; entro la fine della 2a settimana.
vita, la concentrazione di azoto residuo diminuisce al livello osservato negli adulti. La quantità di azoto escreto dai reni aumenta durante i primi 3 giorni di vita, dopodiché diminuisce e ricomincia ad aumentare dalla 2a settimana. vita in parallelo con la crescente quantità di cibo.La massima digeribilità dell'azoto nel corpo del bambino si osserva nei bambini nei primi mesi di vita. Il bilancio azotato si avvicina notevolmente all'equilibrio nei primi 3-6 mesi. vita, anche se rimane positivo. L'intensità del metabolismo proteico nei bambini è piuttosto elevata - nei bambini del 1 ° anno di vita, circa 0,9 G proteine per 1 kg peso corporeo al giorno, in 1-3 anni - 0,8 g/kg/ giorni, nei bambini in età prescolare e scolare - 0,7 g/kg/ giorno
Il valore medio del fabbisogno di aminoacidi essenziali, secondo FAO WHO (1985), nei bambini è 6 volte maggiore che negli adulti (un aminoacido essenziale per i bambini di età inferiore a 3 mesi è la cistina e fino a 5 anni - e istidina). Più attivamente che negli adulti, i processi di transaminazione degli amminoacidi procedono nei bambini. Tuttavia, nei primi giorni di vita nei neonati, a causa dell'attività relativamente bassa di alcuni enzimi, si notano iperaminoacidemia e aminoaciduria fisiologica a causa dell'immaturità funzionale dei reni. Nei bambini prematuri, inoltre, esiste un'aminoaciduria di tipo sovraccarico, tk. il contenuto di aminoacidi liberi nel plasma del loro sangue è più alto che nei bambini a termine. Nella prima settimana di vita, l'azoto amminoacidico costituisce il 3-4% dell'azoto urinario totale (secondo alcune fonti, fino al 10%), e solo entro la fine del 1° anno di vita il suo contenuto relativo diminuisce a 1%. Nei bambini del 1 ° anno di vita, l'escrezione di aminoacidi per 1 kg il peso corporeo raggiunge i valori della loro escrezione in un adulto, l'escrezione di azoto aminoacidico, raggiungendo nei neonati 10 mg/kg peso corporeo, nel 2° anno di vita raramente supera i 2 mg/kg peso corporeo. Nelle urine dei neonati, il contenuto di taurina, treonina, serina, glicina, alanina, cistina, leucina, tirosina, fenilalanina e lisina è aumentato (rispetto all'urina di un adulto). Nei primi mesi di vita, l'etanolamina e l'omocitrullina si trovano anche nelle urine di un bambino. Nelle urine dei bambini del 1° anno di vita predominano gli aminoacidi prolina e [idro]ossiprolina.
Gli studi sui più importanti componenti azotati dell'urina nei bambini hanno dimostrato che il rapporto tra acido urico, urea e ammoniaca cambia significativamente durante la crescita. Sì, per i primi 3 mesi. la vita è caratterizzata dal più basso contenuto di urea nelle urine (2-3 volte inferiore a quello degli adulti) e dalla più alta escrezione di acido urico. I bambini nei primi tre mesi di vita espellono 28.3 mg/kg peso corporeo di acido urico e adulti - 8,7 mg/kg. L'escrezione relativamente elevata di acido urico nei bambini durante i primi mesi di vita a volte contribuisce allo sviluppo dell'infarto da acido urico dei reni. La quantità di urea nelle urine aumenta nei bambini di età compresa tra 3 e 6 mesi e il contenuto di acido urico diminuisce in questo momento. Il contenuto di ammoniaca nelle urine dei bambini nei primi giorni di vita è piccolo, ma poi aumenta bruscamente e rimane ad un livello elevato per tutto il 1 ° anno di vita.
Una caratteristica di A. o. nei bambini è fisiologica la creatinuria. La creatina si trova nel liquido amniotico; nelle urine, è determinato in quantità superiori al contenuto di creatina nelle urine degli adulti, dal periodo neonatale al periodo della pubertà. L'escrezione giornaliera di creatinina (creatina deidrossilata) aumenta con l'età, mentre allo stesso tempo, all'aumentare del peso corporeo del bambino, diminuisce il contenuto relativo di azoto creatininico nelle urine. La quantità di creatinina escreta nelle urine al giorno nei neonati a termine è di 10-13 mg/kg, nei neonati prematuri 3 mg/kg, negli adulti non supera i 30 mg/kg.
All'atto d'identificazione in una famiglia di violazione congenita E. Bisogno consulenza genetica medica.
Bibliografia: Berezov T.T. e Korovkin B.F. Chimica biologica, p. 431, M., 1982; Veltishchev Yu.E. e altri Metabolismo nei bambini, p. 53, M., 1983; Dudel J. et al.Fisiologia umana, trans. dall'inglese, volumi 1-4, M., 1985; Zilva J.F. e Pannell PR Chimica clinica nella diagnosi e nel trattamento, trad. dall'inglese, pag. 298, 398, M., 1988; Kon R.M. e Roy K.S. Diagnosi precoce delle malattie metaboliche, trad. dall'inglese, pag. 211, M., 1986; Metodi di ricerca di laboratorio in clinica, ed. V.V. Menshikov, p. 222, M., 1987; Lehninger A. Fondamenti di biochimica, trad. dall'inglese, volume 2, M., 1985; Mazurin A.V. e Vorontsov I.M. Propedeutica delle malattie infantili, p. 322, M., 1985; Guida alla Pediatria, ed. ed. UE Berman e V.K. Vaughan, trad. dall'inglese, libro. 2, pag. 337 VI, 1987; Strayer L. Biochimica, trad. dall'inglese, vol.2, p. 233, M., 1985.
Domanda completata
metabolismo dell'azoto
metabolismo dell'azoto- un insieme di trasformazioni chimiche, reazioni di sintesi e decomposizione di composti azotati nel corpo; componente del metabolismo e dell'energia. Il concetto di "metabolismo dell'azoto" comprende il metabolismo proteico (un insieme di trasformazioni chimiche nel corpo delle proteine e dei loro prodotti metabolici), nonché lo scambio di peptidi, aminoacidi, acidi nucleici, nucleotidi, basi azotate, amminozuccheri (cfr. carboidrati), contenente azoto lipidi, vitamine, ormoni e altri composti contenenti azoto.
L'organismo degli animali e dell'uomo riceve azoto digeribile dal cibo, in cui la principale fonte di composti azotati sono le proteine di origine animale e vegetale. Il fattore principale per mantenere l'equilibrio dell'azoto - lo stato di AA, in cui la quantità di azoto in entrata e in uscita è la stessa - è un'adeguata assunzione di proteine dal cibo. In URSS, la norma giornaliera delle proteine nella dieta di un adulto è pari a 100 G, o 16 G azoto proteico, con un dispendio energetico di 2500 kcal. Il bilancio azotato (la differenza tra la quantità di azoto che entra nel corpo con il cibo e la quantità di azoto espulso dal corpo con urina, feci e sudore) è un indicatore dell'intensità di A. o. nell'organismo. La fame o un'alimentazione azotata insufficiente portano a un bilancio azotato negativo, o carenza di azoto, in cui la quantità di azoto escreta dal corpo supera la quantità di azoto che entra nel corpo con il cibo. Un bilancio azotato positivo, in cui la quantità di azoto introdotta con il cibo supera la quantità di azoto escreto dal corpo, si osserva durante il periodo di crescita corporea, durante i processi di rigenerazione dei tessuti, ecc. La condizione di A. circa. dipende in gran parte dalla qualità delle proteine alimentari, che a sua volta è determinata dalla sua composizione aminoacidica e, soprattutto, dalla presenza di aminoacidi essenziali.
È generalmente accettato che negli esseri umani e nei vertebrati A. o. inizia con la digestione dei composti azotati del cibo nel tratto gastrointestinale. Nello stomaco, le proteine vengono scomposte con la partecipazione di enzimi proteolitici digestivi. tripsina e gastrixin (cfr Proteolisi ) per formare polipeptidi, oligopeptidi e singoli amminoacidi. Dallo stomaco, la massa alimentare entra nel duodeno e nelle sezioni sottostanti dell'intestino tenue, dove i peptidi subiscono un'ulteriore scissione catalizzata dagli enzimi del succo pancreatico tripsina, chimotripsina e carbossipeptidasi e dagli enzimi del succo intestinale aminopeptidasi e dipeptidasi. Enzimi). Insieme ai peptidi. l'intestino tenue scompone le proteine complesse (p. es., le nucleoproteine) e gli acidi nucleici. Anche la microflora intestinale contribuisce in modo significativo alla scomposizione dei biopolimeri contenenti azoto. Oligopeptidi, amminoacidi, nucleotidi, nucleosidi, ecc. Vengono assorbiti nell'intestino tenue, entrano nel sangue e vengono trasportati con esso in tutto il corpo. Anche le proteine dei tessuti corporei nel processo di costante rinnovamento subiscono la proteolisi sotto l'azione delle protses tissutali (peptidasi e catepsine) e i prodotti di degradazione delle proteine tissutali entrano nel sangue. Gli amminoacidi possono essere utilizzati per la nuova sintesi di proteine e altri composti (basi puriniche e pirimidiniche, nucleotidi, porfirine, ecc.), per produrre energia (ad esempio, attraverso l'inclusione nel ciclo degli acidi tricarbossilici) o possono essere sottoposti a ulteriore degradazione con la formazione di prodotti finali A. O., soggetti a escrezione dal corpo.
Gli aminoacidi che fanno parte delle proteine alimentari sono utilizzati per la sintesi delle proteine degli organi e dei tessuti del corpo. Sono inoltre coinvolti nella formazione di molti altri importanti composti biologici: nucleotidi purinici (glutammina, glicina, acido aspartico) e nucleotidi pirimidinici (glutammina, acido aspartico), serotonina (triptofano), melanina (fenilalpnina, tirosina), istamina (istidina) , adrenalina, noradrenalina, tiramina (tirosina), poliammine (arginina, metionina), colina (metionina), porfirine (glicina), creatina (glicina, arginina, metionina), coenzimi, zuccheri e polisaccaridi, lipidi, ecc. La reazione chimica più importante per l'organismo, alla quale partecipano quasi tutti gli amminoacidi, è la transaminazione, che consiste nel trasferimento enzimatico reversibile del gruppo a-ammino degli amminoacidi all'atomo di carbonio a dei chetoacidi o delle aldeidi. La transaminazione è una reazione fondamentale nella biosintesi degli amminoacidi non essenziali nel corpo. L'attività degli enzimi che catalizzano le reazioni di transaminazione è aminotransferasi - ha un grande valore clinico e diagnostico.
La degradazione degli amminoacidi può procedere attraverso diversi percorsi. La maggior parte degli amminoacidi può subire la decarbossilazione con la partecipazione degli enzimi decarbossilasi per formare ammine primarie, che possono quindi essere ossidate in reazioni catalizzate dalla monoaminossidasi o dalla diammina ossidasi. Quando le ammine biogeniche (istamina, serotonina, tiramina, acido g-amminobutirrico) vengono ossidate dalle ossidasi, si formano le aldeidi, che subiscono ulteriori trasformazioni, e ammoniaca, la principale via di ulteriore metabolismo è la formazione di urea.
Un'altra via principale per la degradazione degli amminoacidi è la deaminazione ossidativa con la formazione di ammoniaca e chetoacidi. La deaminazione diretta degli L-amminoacidi negli animali e nell'uomo procede molto lentamente, ad eccezione dell'acido glutammico, che viene intensamente deaminato con la partecipazione dell'enzima specifico glutammato deidrogenasi. La transaminazione preliminare di quasi tutti gli α-amminoacidi e l'ulteriore deaminazione dell'acido glutammico formatosi in acido α-chetoglutarico e ammoniaca è il meccanismo principale per la deaminazione degli amminoacidi naturali.
L'ammoniaca è il prodotto di vari percorsi di degradazione degli amminoacidi, che possono anche formarsi a seguito del metabolismo di altri composti contenenti azoto (ad esempio, durante la deaminazione dell'adenina, che fa parte della nicotinammide adenina dinucleotide - NAD). Il modo principale di legare e neutralizzare l'ammoniaca tossica negli animali ureotelici (animali in cui il prodotto finale di A. o è l'urea) è il cosiddetto ciclo dell'urea (sinonimo: ciclo dell'ornitina, ciclo di Krebs-Henseleit), che si verifica nel fegato . È una sequenza ciclica di reazioni enzimatiche, a seguito della quale l'urea viene sintetizzata dalla molecola di ammoniaca o dall'azoto ammidico della glutammina, dal gruppo amminico dell'acido aspartico e dall'anidride carbonica. Con un'assunzione giornaliera di 100 G l'escrezione proteica giornaliera di urea dal corpo è di circa 30 G. Nell'uomo e negli animali superiori esiste un altro modo per neutralizzare l'ammoniaca: la sintesi delle ammidi degli acidi dicarbossilici asparagan e glutammina dai corrispondenti amminoacidi. Negli animali uricotelici (rettili, uccelli), il prodotto finale di A. o. è acido urico.
Come risultato della rottura degli acidi nucleici e delle nucleoproteine nel tratto gastrointestinale, si formano nucleotidi e nucleosidi. Oligo- e mono-nucleotidi con la partecipazione di vari enzimi (esterasi, nucleotidasi, nucleosidasi, fosforilasi) vengono quindi convertiti in basi puriniche e pirimidiniche libere.
L'ulteriore percorso di degradazione delle basi puriniche di adenina e guanina consiste nella loro deaminazione idrolitica sotto l'influenza degli enzimi adenasi e guanasi con la formazione rispettivamente di ipoxantina (6-idrossipurina) e xantina (2,6-dioxipurina), che vengono poi convertiti in acido urico in reazioni catalizzate dalla xantina ossidasi. L'acido urico è uno dei prodotti finali di A. o. e il prodotto finale del metabolismo delle purine nell'uomo - viene escreto dal corpo con l'urina. La maggior parte dei mammiferi possiede l'enzima uricase, che catalizza la conversione dell'acido urico in allantoina escreta.
La degradazione delle basi pirimidiniche (uracile, timina) consiste nella loro riduzione con formazione di diidroderivati e successiva idrolisi, a seguito della quale si forma acido b-ureidopropionico dall'uracile e da esso ammoniaca, anidride carbonica e b-alanina, e acido b-amminoisobutirrico da timina, acido, anidride carbonica e ammoniaca. L'anidride carbonica e l'ammoniaca possono essere ulteriormente incluse nell'urea attraverso il ciclo dell'urea e la b-alanina è coinvolta nella sintesi dei più importanti composti biologicamente attivi: i dipeptidi contenenti istidina carnosina (b-alanil-L-istidina) e anserina (b -alanil-N-metil-L-istidina), presente nelle sostanze estrattive dei muscoli scheletrici, nonché nella sintesi dell'acido pantotenico e del coenzima A.
Pertanto, varie trasformazioni dei più importanti composti azotati del corpo sono interconnesse in un unico scambio. Processo complicato A. o. regolato a livello molecolare, cellulare e tissutale. Il regolamento di A. circa. in tutto l'organismo ha lo scopo di adattare l'intensità di A. o. alle mutevoli condizioni dell'ambiente e dell'ambiente interno ed è svolto dal sistema nervoso sia direttamente che agendo sulle ghiandole endocrine.
Negli adulti sani, il contenuto di composti azotati in organi, tessuti e fluidi biologici è a un livello relativamente costante. L'eccesso di azoto dal cibo viene escreto nelle urine e nelle feci e, con una mancanza di azoto nel cibo, il fabbisogno del corpo può essere coperto dall'uso di composti azotati nei tessuti corporei. Allo stesso tempo, la composizione urina cambia a seconda delle caratteristiche E. e bilancio azotato. Normalmente, con una dieta invariata e condizioni ambientali relativamente stabili, una quantità costante di prodotti finali di AA viene escreta dal corpo e lo sviluppo di condizioni patologiche porta al suo brusco cambiamento. Cambiamenti significativi nell'escrezione di composti azotati nelle urine, principalmente nell'escrezione di urea, possono essere osservati anche in assenza di patologia in caso di un cambiamento significativo nella dieta (ad esempio, quando la quantità di proteine consumate viene modificata ), e la concentrazione di azoto residuo (cfr. Azoto residuo ) nel sangue cambia leggermente.
In una ricerca E. è necessario tener conto della composizione quantitativa e qualitativa del cibo assunto, della composizione quantitativa e qualitativa dei composti azotati escreti nelle urine e nelle feci e contenuti nel sangue. Per la ricerca di A. su. utilizzare sostanze azotate etichettate con radionuclidi di azoto, fosforo, carbonio, zolfo, idrogeno, ossigeno e osservare la migrazione dell'etichetta e la sua incorporazione nella composizione dei prodotti finali di A. o. Gli amminoacidi marcati sono ampiamente utilizzati, ad esempio 15 N-glicina, che vengono introdotti nel corpo con il cibo o direttamente nel sangue. Una parte significativa dell'azoto glicina alimentare etichettato viene escreta come urea con l'urina e l'altra parte dell'etichetta entra nelle proteine dei tessuti ed è espulsa dal corpo molto lentamente. Condurre ricerche A. o. necessario per la diagnosi di molte condizioni patologiche e il monitoraggio dell'efficacia del trattamento, nonché lo sviluppo di diete razionali, incl. medicinale (cfr Nutrizione medica ).
Patologia A. o. (fino a molto significativo) provoca carenza di proteine. Può essere causato da malnutrizione generale, carenza prolungata di proteine o aminoacidi essenziali nella dieta, mancanza di carboidrati e grassi che forniscono energia per i processi di biosintesi proteica nel corpo. La carenza proteica può essere dovuta alla predominanza dei processi di degradazione proteica sulla loro sintesi, non solo come risultato di carenza alimentare di proteine e altri nutrienti essenziali, ma anche durante un lavoro muscolare pesante, lesioni, processi infiammatori e distrofici, ischemia, infezione, ustioni, un difetto nella funzione trofica del sistema nervoso sistema, insufficienza di ormoni anabolici (ormone della crescita, ormoni sessuali, insulina), sintesi eccessiva o assunzione eccessiva di ormoni steroidei dall'esterno, ecc. Violazione dell'assorbimento proteico nella patologia del tratto gastrointestinale (evacuazione accelerata del cibo dallo stomaco, condizioni ipo e anacide, blocco del dotto escretore del pancreas, indebolimento della funzione secretoria e aumento della motilità dell'intestino tenue nell'enterite e enterocolite, assorbimento alterato nell'intestino tenue, ecc.) può anche portare a carenza di proteine. La carenza di proteine porta alla discordanza A. o. ed è caratterizzato da un pronunciato bilancio azotato negativo.
Sono noti casi di violazione della sintesi di alcune proteine (vedi. Immunopatologia, Fermentopatie), così come la sintesi geneticamente determinata di proteine anormali, ad esempio con emoglobinopatie, mieloma multiplo (cfr Emoblastosi paraproteinemiche ) e così via.
La patologia di A. o., che consiste in una violazione del metabolismo degli aminoacidi, è spesso associata ad anomalie nel processo di transaminazione: una diminuzione dell'attività delle aminotransferasi durante l'ipo- o l'avitaminosi B 6, una violazione della sintesi di questi enzimi, una mancanza di chetoacidi per la transaminazione dovuta all'inibizione del ciclo dell'acido tricarbossilico durante l'ipossia e il diabete da zucchero, ecc. Una diminuzione dell'intensità della transaminazione porta all'inibizione della deaminazione dell'acido glutammico e questo, a sua volta, ad un aumento della proporzione di azoto amminoacidico nella composizione dell'azoto residuo nel sangue (iperaminoacidemia), iperazotemia generale e aminoaciduria. Iperaminoacidemia, aminoaciduria e azotemia generale sono caratteristiche di molti tipi di patologia di A.. Con estesi danni al fegato e altre condizioni associate a una massiccia disgregazione proteica nel corpo, i processi di deaminazione degli amminoacidi e la formazione di urea vengono interrotti in modo tale che la concentrazione di azoto residuo e il contenuto di azoto amminoacidico in esso aumentano sullo sfondo di una diminuzione del contenuto relativo di azoto ureico nell'azoto residuo (la cosiddetta produzione di azotemia).
L'azotemia di produzione è solitamente accompagnata dall'escrezione di aminoacidi in eccesso nelle urine, poiché anche in caso di normale funzionamento dei reni, la filtrazione degli aminoacidi nei glomeruli renali è più intensa del loro riassorbimento nei tubuli. Malattie renali, ostruzione delle vie urinarie, alterata circolazione renale portano allo sviluppo di azotemia da ritenzione, accompagnata da un aumento della concentrazione di azoto residuo nel sangue dovuto ad un aumento del contenuto di urea nel sangue (vedi. insufficienza renale ). Ferite estese, gravi ustioni, infezioni, danni alle ossa tubolari, al midollo spinale e al cervello, ipotiroidismo, malattia di Itsenko-Cushing e molte altre gravi malattie sono accompagnate da aminoaciduria. È anche caratteristico delle condizioni patologiche che si verificano con processi di riassorbimento alterati nei tubuli renali: malattia di Wilson-Konovalov (vedi. Distrofia epatocerebrale ), Nefronoftisi Fanconi (cfr. Malattie simili al rachitismo ) e altri Queste malattie sono tra i numerosi disturbi geneticamente determinati di A. o. La violazione selettiva del riassorbimento della cistina e della cistinuria con un disturbo generalizzato del metabolismo della cistina sullo sfondo dell'aminoaciduria generale accompagna la cosiddetta cistinosi. In questa malattia, i cristalli di cistina si depositano nelle cellule del sistema reticoloendoteliale. malattia ereditaria fenilchetonuria caratterizzato da una violazione della conversione della fenilalanina in tirosina a seguito di una deficienza geneticamente determinata dell'enzima fenilalanina - 4-idrossilasi, che provoca l'accumulo nel sangue e nelle urine di fenilalanina non convertita e dei suoi prodotti metabolici - acido fenilpiruvico e fenilacetico. La violazione delle trasformazioni di questi composti è anche caratteristica dell'epatite virale.
Tirosinemia, tirosinuria e tironosi si notano nella leucemia, nelle malattie diffuse del tessuto connettivo (collagenosi) e in altre condizioni patologiche. Si sviluppano a causa della compromissione della transaminazione della tirosina. Un'anomalia congenita delle trasformazioni ossidative della tirosina è alla base dell'alcaptonuria, in cui un metabolita non convertito di questo amminoacido, l'acido omogentisico, si accumula nelle urine. Disturbi del metabolismo dei pigmenti nell'ipocorticismo (vedi. ghiandole surrenali ) sono associati all'inibizione della conversione della tirosina in melanina dovuta all'inibizione dell'enzima tirosinasi (la completa perdita della sintesi di questo pigmento è caratteristica di un'anomalia congenita della pigmentazione - albinismo).
Nell'epatite cronica, diabete mellito, leucemia acuta, leucemia mielo- e linfocitica cronica, linfogranulomatosi, reumatismi e sclerodermia, il metabolismo del triptofano è disturbato e i suoi metaboliti 3-idrossicinurenina, acido xanturenico e 3-idrossiantranilico, che hanno proprietà tossiche, si accumulano nel sangue . Alla patologia di A. su. includere anche condizioni associate a una violazione dell'escrezione di creatinina da parte dei reni e del suo accumulo nel sangue. Un aumento dell'escrezione di creatinina accompagna l'iperfunzione della ghiandola tiroidea e una diminuzione dell'escrezione di creatinina con un aumento dell'escrezione di creatina è l'ipotiroidismo.
Con una massiccia rottura delle strutture cellulari (fame, lavoro muscolare pesante, infezioni, ecc.), Si nota un aumento patologico della concentrazione di azoto residuo dovuto ad un aumento del contenuto relativo di azoto dell'acido urico in esso (normalmente, la concentrazione di acido urico nel sangue non supera - 0,4 mmol/l).
Nella vecchiaia, l'intensità e il volume della sintesi proteica diminuiscono a causa dell'inibizione diretta della funzione biosintetica dell'organismo e dell'indebolimento della sua capacità di assorbire gli aminoacidi alimentari; si sviluppa un bilancio azotato negativo. I disturbi del metabolismo delle purine negli anziani portano all'accumulo e alla deposizione di sali di acido urico - urati nei muscoli, nelle articolazioni e nella cartilagine. Correzione di disturbi E. in età avanzata può essere effettuata attraverso diete speciali contenenti proteine animali di alta qualità, vitamine e oligoelementi, con un contenuto limitato di purine.
Il metabolismo dell'azoto nei bambini si distingue per una serie di caratteristiche, in particolare un bilancio azotato positivo come condizione necessaria per la crescita. L'intensità dei processi di A. o. durante la crescita del bambino subisce cambiamenti, particolarmente pronunciati nei neonati e nei bambini piccoli. Durante i primi 3 giorni di vita, il bilancio azotato è negativo, il che si spiega con un apporto insufficiente di proteine dal cibo. Durante questo periodo viene rilevato un aumento transitorio della concentrazione di azoto residuo nel sangue (la cosiddetta azotemia fisiologica), che talvolta raggiunge i 70 mmol/l; entro la fine della 2a settimana.
vita, la concentrazione di azoto residuo diminuisce al livello osservato negli adulti. La quantità di azoto escreto dai reni aumenta durante i primi 3 giorni di vita, dopodiché diminuisce e ricomincia ad aumentare dalla 2a settimana. vita in parallelo con la crescente quantità di cibo.
La massima digeribilità dell'azoto nel corpo del bambino si osserva nei bambini nei primi mesi di vita. Il bilancio azotato si avvicina notevolmente all'equilibrio nei primi 3-6 mesi. vita, anche se rimane positivo. L'intensità del metabolismo proteico nei bambini è piuttosto elevata - nei bambini del 1 ° anno di vita, circa 0,9 G proteine per 1 kg peso corporeo al giorno, in 1-3 anni - 0,8 g/kg/ giorni, nei bambini in età prescolare e scolare - 0,7 g/kg/ giorno
Il valore medio del fabbisogno di aminoacidi essenziali, secondo FAO WHO (1985), nei bambini è 6 volte maggiore che negli adulti (un aminoacido essenziale per i bambini di età inferiore a 3 mesi è la cistina e fino a 5 anni - e istidina). Più attivamente che negli adulti, i processi di transaminazione degli amminoacidi procedono nei bambini. Tuttavia, nei primi giorni di vita nei neonati, a causa dell'attività relativamente bassa di alcuni enzimi, si notano iperaminoacidemia e aminoaciduria fisiologica a causa dell'immaturità funzionale dei reni. Nei bambini prematuri, inoltre, esiste un'aminoaciduria di tipo sovraccarico, tk. il contenuto di aminoacidi liberi nel plasma del loro sangue è più alto che nei bambini a termine. Nella prima settimana di vita, l'azoto amminoacidico costituisce il 3-4% dell'azoto urinario totale (secondo alcune fonti, fino al 10%), e solo entro la fine del 1° anno di vita il suo contenuto relativo diminuisce a 1%. Nei bambini del 1 ° anno di vita, l'escrezione di aminoacidi per 1 kg il peso corporeo raggiunge i valori della loro escrezione in un adulto, l'escrezione di azoto aminoacidico, raggiungendo nei neonati 10 mg/kg peso corporeo, nel 2° anno di vita raramente supera i 2 mg/kg peso corporeo. Nelle urine dei neonati, il contenuto di taurina, treonina, serina, glicina, alanina, cistina, leucina, tirosina, fenilalanina e lisina è aumentato (rispetto all'urina di un adulto). Nei primi mesi di vita, l'etanolamina e l'omocitrullina si trovano anche nelle urine di un bambino. Nelle urine dei bambini del 1° anno di vita predominano gli aminoacidi prolina e [idro]ossiprolina.
Gli studi sui più importanti componenti azotati dell'urina nei bambini hanno dimostrato che il rapporto tra acido urico, urea e ammoniaca cambia significativamente durante la crescita. Sì, per i primi 3 mesi. la vita è caratterizzata dal più basso contenuto di urea nelle urine (2-3 volte inferiore a quello degli adulti) e dalla più alta escrezione di acido urico. I bambini nei primi tre mesi di vita espellono 28.3 mg/kg peso corporeo di acido urico e adulti - 8,7 mg/kg. L'escrezione relativamente elevata di acido urico nei bambini durante i primi mesi di vita a volte contribuisce allo sviluppo dell'infarto da acido urico dei reni. La quantità di urea nelle urine aumenta nei bambini di età compresa tra 3 e 6 mesi e il contenuto di acido urico diminuisce in questo momento. Il contenuto di ammoniaca nelle urine dei bambini nei primi giorni di vita è piccolo, ma poi aumenta bruscamente e rimane ad un livello elevato per tutto il 1 ° anno di vita.
Una caratteristica di A. o. nei bambini è fisiologica la creatinuria. La creatina si trova nel liquido amniotico; nelle urine, è determinato in quantità superiori al contenuto di creatina nelle urine degli adulti, dal periodo neonatale al periodo della pubertà. L'escrezione giornaliera di creatinina (creatina deidrossilata) aumenta con l'età, mentre allo stesso tempo, all'aumentare del peso corporeo del bambino, diminuisce il contenuto relativo di azoto creatininico nelle urine. La quantità di creatinina escreta nelle urine al giorno nei neonati a termine è di 10-13 mg/kg, nei neonati prematuri 3 mg/kg, negli adulti non supera i 30 mg/kg.
All'atto d'identificazione in una famiglia di violazione congenita E. Bisogno
Breve domanda
Isolamento dei prodotti finali del metabolismo dell'azoto
L'acido urico è uno dei prodotti finali più importanti del metabolismo dell'azoto nell'uomo. Normalmente, la sua concentrazione nel siero del sangue negli uomini è 0,27-0,48 mmol-l1, nelle donne 0,18-0,38 mmol-l-1; l'escrezione urinaria giornaliera varia da 2,3 a 4,5 mmol (400-750 mg). Gli esseri umani espellono l'acido urico e molti mammiferi hanno l'enzima uricase, che ossida l'acido urico in allantoina. Nel corpo di una persona sana al giorno, la formazione e l'escrezione di acido urico varia da 500 a 700 mg. La maggior parte dell'acido urico (fino all'80%) si forma a seguito del metabolismo degli acidi nucleici endogeni, solo il 20% circa è associato alle purine dal cibo. I reni espellono circa 500 mg di acido urico al giorno, 200 mg vengono rimossi attraverso il tratto gastrointestinale.
Proteinuria funzionale. Proteinuria funzionale, i cui esatti processi di occorrenza non sono stati determinati, includono quelli relativi alla posizione verticale del corpo, idiopatica non permanente, escrezione di proteine nelle urine di ceppo, aspetto febbrile di proteine nelle urine ed escrezione di proteine nelle urine dell'obesità.
La proteinuria ortostatica è caratterizzata dalla comparsa di un polipeptide nelle urine durante una prolungata inattività o stimolazione, con la sua rapida scomparsa quando la postura del corpo cambia in perpendicolare. L'aspetto delle proteine nelle urine nella maggior parte dei casi non supera un g / die, è glomerulare e non selettivo, la procedura per il suo aspetto non è chiara. Più spesso si nota nell'adolescenza, nella metà dei pazienti guarisce dopo un po '. Il meccanismo di formazione è forse associato a una risposta anormalmente intensificata della circolazione renale a un cambiamento nella posizione del tronco.
La definizione di proteinuria ortostatica è data dalla combinazione delle seguenti condizioni:
Età dei pazienti 13-20 anni;
Un tipo chiuso di proteine nelle urine, l'assenza di altri segni di danno renale (ristrutturazione del sedimento urinario, aumento della pressione che il sangue nell'arteria esercita sulla sua parete, modificazioni dei vasi della superficie interna del bulbo oculare);
Solo un decorso ortostatico della comparsa di proteine nelle urine, quando negli studi sull'urina prelevati dopo che il soggetto era in posizione supina (compresa la mattina successiva prima di alzarsi dal letto), non ci sono proteine.
Per dimostrare questa diagnosi, è necessario eseguire un test verticale. Per fare questo, l'urina viene raccolta al mattino prima di alzarsi dal letto, quindi dopo essere stati in posizione perpendicolare per un po' di tempo (muovendosi con un bastoncino dietro la schiena per dispiegare la colonna vertebrale). La diagnostica fornisce risultati ancora più accurati quando la porzione mattutina (notte) dell'urina si fonde (poiché l'urina residua è possibile nella vescica urinaria) e la porzione iniziale viene selezionata dopo una breve presenza del paziente in posizione supina.
In giovane età, a sua volta, è possibile la comparsa primaria intermittente di proteine nelle urine, che si instaura in individui sani durante una visita medica e scompare durante i successivi esami delle urine.
La proteinuria da tensione viene rilevata nel venti percento degli individui sani (anche atleti) dopo un intenso sforzo fisico. La proteina viene rilevata nella porzione iniziale di urina preparata. escrezione di proteine nelle urine di natura associata alla patologia dei tubuli. Si presume che l'algoritmo per la comparsa delle proteine nelle urine sia combinato con la ricombinazione del flusso sanguigno e la relativa ischemia delle parti prossimali del nefrone.
L'aspetto febbrile delle proteine nelle urine si verifica in condizioni di caldo intenso, in particolare nei bambini e nelle persone senili. L'escrezione febbrile di proteine nelle urine ha un decorso prevalentemente glomerulare. I processi di questo tipo di proteinuria sono poco conosciuti, si sta studiando la possibile importanza di un aumento della filtrazione glomerulare insieme a un danno di breve durata al filtro glomerulare da parte di complessi protettivi.
Isolamento delle proteine nelle urine in sovrappeso patologico. L'escrezione di proteine nelle urine è spesso osservata con deposizione anormale di grasso nel corpo. (peso corporeo superiore a 115 chilogrammi). Secondo J.P.Domfeld (1989), tra mille pazienti con deposizione patologica di grasso nel corpo. a 420 è stata diagnosticata l'escrezione di proteine nelle urine senza degenerazione dei fanghi urinari; i precedenti di una sindrome nephrotic sono mostrati anche. Si presume che la causa principale della formazione di tale proteinuria sia la distorsione della circolazione sanguigna dell'accumulo di capillari altamente fenestrati (aumento della pressione nel gruppo di capillari del corpuscolo renale, aumento della velocità di filtrazione), associata ad un aumento della sovrappeso patologico. la concentrazione di un ormone polipeptidico prodotto dal rene a causa di una diminuzione della pressione sanguigna e dell'ipertensione, che diminuisce durante il digiuno. Con la perdita di peso, così come durante il trattamento con ACE-inibitori, l'escrezione proteica urinaria può diminuire, oltre che essere persa.
Inoltre, la proteinuria può avere un esordio extrarenale. In presenza di leucociti nel test delle urine e in particolare la comparsa di sangue nelle urine, una reazione di conferma al polipeptide può essere il risultato della rottura delle cellule del sangue durante la permanenza prolungata dell'urina, in questa situazione, la comparsa di proteine in l'urina superiore a 0,3 grammi / giorno sembra anormale. I test polipeptidici sedimentari possono dare risultati falsi positivi in presenza di materiali di contrasto contenenti iodio nelle urine, un numero considerevole di preparazioni di penicillinum simili, nonché una sostanza medicinale del gruppo di antibiotici beta-lattamici semisintetici, prodotti metabolici di preparazioni di sulfanilamide.
Informazioni simili.
- In contatto con 0
- Google+ 0
- OK 0
- Facebook 0






