- una condizione ereditaria geneticamente eterogenea caratterizzata da una violazione della struttura e della funzionalità di alcuni canali ionici dei cardiomiociti. La gravità delle manifestazioni della patologia varia in un intervallo molto ampio: da un decorso praticamente asintomatico (vengono rilevati solo segni elettrocardiologici) a grave sordità, svenimenti e aritmie. La definizione di sindrome del QT lungo si basa su dati provenienti da studi elettrocardiologici e analisi genetiche molecolari. Il trattamento dipende dalla forma della patologia e può includere l'assunzione continua o continua di beta-bloccanti, preparati di magnesio e potassio, nonché l'installazione di un defibrillatore-cardioverter.

informazioni generali
La sindrome del QT lungo è un gruppo di disturbi cardiaci di natura genetica, in cui il passaggio delle correnti ioniche nei cardiomiociti viene interrotto, il che può portare ad aritmie, svenimenti e morte cardiaca improvvisa. Per la prima volta, una tale condizione fu identificata nel 1957 dai medici norvegesi A. Jervell e F. Lange-Nielsen, che descrissero una combinazione di sordità congenita, attacchi sincopali e prolungamento dell'intervallo QT in un paziente. Un po' più tardi, nel 1962-64, sintomi simili furono riscontrati in pazienti con udito normale - tali casi furono descritti indipendentemente da C. Romano e O. Ward.
Questo, così come ulteriori scoperte, hanno determinato la divisione della sindrome del QT lungo in due varianti cliniche: Romano-Ward e Jervell-Lange-Nielsen. La prima è ereditata con meccanismo autosomico dominante, la sua frequenza nella popolazione è di 1 caso ogni 5.000 abitanti. L'incidenza della sindrome del QT lungo di tipo Jervell-Lange-Nielsen varia da 1 a 6:1.000.000, è caratterizzata da una trasmissione autosomica dominante e da manifestazioni più pronunciate. Secondo alcuni rapporti, tutte le forme di sindrome del QT lungo sono responsabili di un terzo dei casi di morte cardiaca improvvisa e di circa il 20% di morte improvvisa infantile.
Cause e classificazione
Attualmente sono stati identificati 12 geni, le cui mutazioni portano allo sviluppo della sindrome del QT lungo, che codificano per alcune proteine che fanno parte dei canali ionici dei cardiomiociti responsabili della corrente ionica di sodio o potassio. È stato anche possibile trovare le ragioni delle differenze nel decorso clinico di questa malattia. La sindrome di Romano-Ward autosomica dominante è causata da una mutazione in un solo gene e pertanto può essere asintomatica o almeno senza perdita dell'udito. Nel tipo Jervell-Lange-Nielsen c'è un difetto in due geni: questa variante, oltre ai sintomi cardiaci, è sempre accompagnata da sordità neurosensoriale bilaterale. Ad oggi, le mutazioni dei cui geni sono noti per causare lo sviluppo della sindrome del QT lungo:
- Sindrome del QT lungo di tipo 1 (LQT1) a causa di una mutazione del gene KCNQ1 situato sull'11° cromosoma. Difetti in questo gene vengono spesso rilevati in presenza di questa malattia. Codifica la sequenza della subunità alfa di una delle varietà di canali del potassio nei cardiomiociti (lKs)
- Sindrome del QT lungo di tipo 2 (LQT2)è causata da difetti nel gene KCNH2, che si trova sul 7° cromosoma e codifica la sequenza aminoacidica della proteina, la subunità alfa di un altro tipo di canali del potassio (lKr).
- Sindrome del QT lungo di tipo 3 (LQT3) a causa di una mutazione del gene SCN5A localizzato sul 3° cromosoma. A differenza delle precedenti varianti della patologia, ciò interrompe il lavoro dei canali del sodio dei cardiomiociti, poiché questo gene codifica la sequenza della subunità alfa del canale del sodio (lNa).
- Sindrome del QT lungo di tipo 4 (LQT4)- una variante piuttosto rara della malattia causata da una mutazione del gene ANK2, che si trova sul 4° cromosoma. Il prodotto della sua espressione è la proteina anchirina B, che nel corpo umano è coinvolta nella stabilizzazione della struttura dei microtubuli dei miociti, ed è anche secreta nelle cellule neurogliali e retiniche.
- Sindrome del QT lungo di tipo 5 (LQT5)- un tipo di malattia causata da un difetto nel gene KCNE1, localizzato sul 21° cromosoma. Codifica una delle proteine dei canali ionici, la subunità beta dei canali del potassio di tipo lKs.
- Sindrome del QT lungo di tipo 6 (LQT6) causata da una mutazione nel gene KCNE2, anch'esso situato sul 21° cromosoma. Il suo prodotto di espressione è la subunità beta dei canali del potassio di tipo lKr.
- Sindrome del QT lungo tipo 7(LQT7, un altro nome - sindrome di Andersen, in onore del pediatra E. D. Andersen, che descrisse questa malattia negli anni '70) è causata da un difetto nel gene KCNJ2, che è localizzato sul 17o cromosoma. Come nel caso delle precedenti varianti della patologia, questo gene codifica per una delle catene proteiche dei canali del potassio.
- Sindrome del QT lungo tipo 8(LQT8, un altro nome è sindrome di Timothy, in onore di K. Timothy, che descrisse questa malattia) è causata da una mutazione del gene CACNA1C, che si trova sul 12° cromosoma. Questo gene codifica per la subunità alfa-1 del canale del calcio di tipo L.
- Sindrome del QT lungo di tipo 9 (LQT9) a causa di un difetto nel gene CAV3 situato sul 3° cromosoma. Il prodotto della sua espressione è la proteina caveolina 3, coinvolta nella formazione di numerose strutture sulla superficie dei cardiomiociti.
- Sindrome del QT lungo di tipo 10 (LQT10)- la causa di questo tipo di malattia risiede nella mutazione del gene SCN4B, che si trova sull'11° cromosoma ed è responsabile della sequenza aminoacidica della subunità beta dei canali del sodio.
- Sindrome del QT lungo di tipo 11 (LQT11) causata da difetti nel gene AKAP9 situato sul cromosoma 7. Codifica una proteina specifica: l'A-chinasi dei centrosomi e il complesso del Golgi. Le funzioni di questa proteina non sono ancora ben comprese.
- Sindrome del QT lungo di tipo 12 (LQT12) a causa di una mutazione del gene SNTA1 localizzato sul 20° cromosoma. Codifica la subunità alfa-1 della proteina sintrofina, che è coinvolta nella regolazione dell'attività dei canali del sodio nei cardiomiociti.
Nonostante l'ampia diversità genetica della sindrome del QT lungo, i collegamenti generali della sua patogenesi sono generalmente gli stessi per ciascuna delle forme. Questa malattia appartiene al gruppo delle canalopatie perché è causata da disturbi nella struttura di alcuni canali ionici. Di conseguenza, i processi di ripolarizzazione miocardica si verificano in modo non uniforme e non simultaneo in diverse parti dei ventricoli, il che provoca un prolungamento dell'intervallo QT. Inoltre, la sensibilità del miocardio agli influssi del sistema nervoso simpatico aumenta in modo significativo, il che provoca frequenti tachiaritmie che possono portare a fibrillazione ventricolare pericolosa per la vita. Allo stesso tempo, diversi tipi genetici della sindrome del QT lungo hanno una sensibilità diversa a determinati influssi. Ad esempio, la LQT1 è caratterizzata da crisi sincopali e aritmie durante l'esercizio, con la LQT2 si osservano manifestazioni simili con suoni forti e acuti, per la LQT3, al contrario, lo sviluppo di aritmie e fibrillazioni in uno stato calmo (ad esempio, nel sonno ) è più caratteristico.
Sintomi del QT lungo
Le manifestazioni della sindrome del QT lungo sono piuttosto diverse. Con una forma clinica più grave di Jervell-Lange-Nielsen, i pazienti presentano sordità, svenimenti frequenti, vertigini e debolezza. Inoltre, in alcuni casi, in questa condizione si registrano convulsioni convulsive di tipo epilettoide, che spesso portano a diagnosi e trattamenti errati. Secondo alcuni genetisti, dal 10 al 25% dei pazienti affetti dalla sindrome del QT lungo ricevono un trattamento sbagliato e sviluppano morte cardiaca improvvisa o morte infantile. L'insorgenza di tachiaritmie e condizioni sincopali dipende da influenze esterne: ad esempio, con LQT1 ciò può verificarsi sullo sfondo dell'attività fisica, con LQT2, la perdita di coscienza e la fibrillazione ventricolare possono verificarsi a causa di suoni acuti e forti.
Una forma più lieve di sindrome del QT lungo (tipo Romano-Ward) è caratterizzata da sincope transitoria (sincope) e rari attacchi di tachiaritmia, ma non vi è perdita dell'udito. In alcuni casi, questa forma della malattia non si manifesta in alcun modo, ad eccezione dei dati elettrocardiografici, ed è un riscontro accidentale durante una visita medica. Tuttavia, anche con questo decorso della sindrome del QT lungo, il rischio di morte cardiaca improvvisa dovuta alla fibrillazione ventricolare è molte volte superiore rispetto a una persona sana. Pertanto, questo tipo di patologia richiede uno studio attento e un trattamento preventivo.
Diagnostica
La diagnosi della sindrome del QT lungo viene effettuata sulla base di uno studio dell'anamnesi del paziente, di studi elettrocardiologici e di genetica molecolare. Interrogando il paziente si riscontrano spesso episodi di svenimento, vertigini, palpitazioni, ma nelle forme lievi di patologia potrebbero non esserlo. A volte manifestazioni simili si verificano in uno dei parenti del paziente, il che indica la natura familiare della malattia.
Con qualsiasi forma di sindrome del QT lungo, verranno rilevati cambiamenti nell'ECG: un aumento dell'intervallo QT a 0,6 secondi o più, è possibile un aumento dell'ampiezza dell'onda T. La combinazione di tali segni ECG con sordità congenita indica la presenza della sindrome di Jervell-Lange-Nielsen. Inoltre, il monitoraggio Holter del lavoro cardiaco durante il giorno è spesso necessario per identificare possibili attacchi di tachiaritmie. La definizione della sindrome del QT lungo con i metodi della genetica moderna è attualmente possibile per quasi tutti i tipi genetici di questa malattia.
Trattamento della sindrome del QT lungo
La terapia per la sindrome del QT lungo è piuttosto complicata, molti esperti raccomandano alcuni schemi per questa malattia e ne rifiutano altri, ma non esiste un protocollo unico per il trattamento di questa patologia. I beta-bloccanti sono considerati farmaci universali, che riducono il rischio di sviluppare tachiaritmie e fibrillazioni, oltre a ridurre il grado di effetti simpatici sul miocardio, ma sono inefficaci nella LQT3. Nel caso della sindrome del QT lungo di tipo 3, è più ragionevole utilizzare farmaci antiaritmici di classe B1. Queste caratteristiche del trattamento della malattia aumentano la necessità di diagnostica genetica molecolare per determinare il tipo di patologia. In caso di frequenti attacchi di tachiaritmie e di alto rischio di sviluppare fibrillazione, si consiglia l'impianto di un pacemaker o di un defibrillatore cardioverter.
Previsione
La prognosi della sindrome del QT lungo, secondo la maggior parte degli esperti, è incerta, poiché questa malattia è caratterizzata da un'ampia gamma di sintomi. Inoltre, l'assenza di manifestazioni patologiche, ad eccezione dei dati elettrocardiografici, non garantisce lo sviluppo improvviso della fibrillazione ventricolare fatale sotto l'influenza di fattori esterni o interni. Se viene identificata la sindrome del QT lungo, è necessario un esame cardiaco approfondito e la determinazione genetica del tipo di malattia. Sulla base dei dati ottenuti, viene sviluppato un regime di trattamento per ridurre la probabilità di morte cardiaca improvvisa o viene presa la decisione di impiantare un pacemaker.
Fai un test online (esame) su questo argomento...
Intervallo QT(sistole elettrica ventricolare) - il tempo dall'inizio del complesso QRT alla fine dell'onda T. L'intervallo QT dipende dal sesso, dall'età (nei bambini l'intervallo è più breve) e dalla frequenza cardiaca.
Normalmente, l'intervallo QT è 0,35-0,44 s (17,5-22 cellule). L'intervallo QT è un valore costante per la frequenza del ritmo (separatamente per uomini e donne). Esistono tabelle speciali che presentano gli standard QT per un dato sesso e frequenza del ritmo. Se il risultato dell'ECG supera 0,05 secondi (2,5 celle) del valore della tabella, allora si parla di allungamento della sistole elettrica dei ventricoli, che è un segno caratteristico della cardiosclerosi.
Secondo la formula di Bazett è possibile determinare se l'intervallo QT in un dato paziente è normale o patologico (l'intervallo QT è considerato patologico quando il valore supera 0,42):
QT=QT(misurato dall'ECG, sec) / √(R-R)(intervallo, misurato dall'ECG, tra due onde R adiacenti, sec)


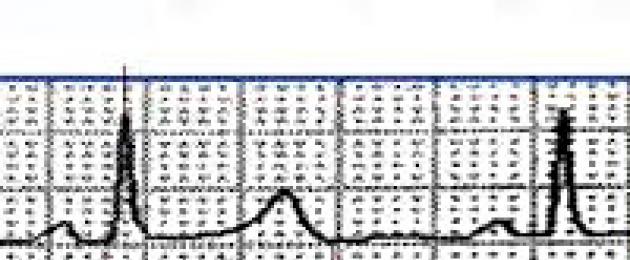
Ad esempio, il valore QT calcolato per l'ECG mostrato a destra (calcolato dalla derivazione standard II:
- L'intervallo QT è di 17 cellule (0,34 secondi).
- La distanza tra due onde R è di 46 celle (0,92 secondi).
- La radice quadrata di 0,92 = 0,96.
- QT=0,34/0,96=0,35
Sono stati identificati i geni responsabili dello sviluppo della malattia, studiata la funzione dei cardiomiociti a livello molecolare e le manifestazioni cliniche. La decifrazione delle mutazioni nei geni che codificano gli elementi strutturali proteici di alcuni canali ionici ha permesso di stabilire una chiara relazione tra genotipo e fenotipo.
Fisiopatologia
La sindrome dell'intervallo OT lungo si sviluppa a causa del prolungamento del periodo di ripolarizzazione dei cardiomiociti ventricolari, che si manifesta con un prolungamento dell'intervallo OT sull'ECG, predispone alla comparsa di aritmie ventricolari sotto forma di tachicardia del tipo "piroetta" , fibrillazione ventricolare, morte cardiaca improvvisa. Il potenziale d'azione di un cardiomiocita è generato dal lavoro coordinato di almeno 10 canali ionici (che trasportano principalmente ioni sodio, calcio e potassio attraverso la membrana cellulare). I disturbi funzionali di uno qualsiasi di questi meccanismi (acquisiti o determinati geneticamente), che portano ad un aumento delle correnti di depolarizzazione o ad un indebolimento del processo di ripolarizzazione, possono causare lo sviluppo della sindrome.
Forma congenita della sindrome
Due forme ereditarie di questa patologia sono ben studiate. La più comune è la sindrome di Romano-Ward (una malattia autosomica dominante con penetranza variabile che non presenta altre caratteristiche fenotipiche) e la meno comune sindrome di Jervell-Lange-Nielsen, una malattia autosomica recessiva associata alla sordità. La moderna classificazione genetica ha ora sostituito questi eponimi. Sono stati identificati sei loci cromosomici (LQTS1-6), che codificano sei geni responsabili dell'insorgenza della patologia. Ciascuna delle sindromi genetiche presenta anche manifestazioni cliniche caratteristiche.
Esiste una connessione tra forme congenite e acquisite. I portatori dell'anomalia genetica potrebbero non mostrare segni elettrocardiografici caratteristici, ma quando assumono farmaci che prolungano l'intervallo QT, come l'eritromicina, tali persone possono sviluppare torsioni di punta e causare morte improvvisa.
Forma acquisita della sindrome
Manifestazioni cliniche
Un segno caratteristico della sindrome da prolungamento dell'intervallo OT è lo svenimento ripetuto, provocato da stress emotivo o fisico. Allo stesso tempo si osserva un'aritmia di tipo piroetta, che è spesso preceduta da cicli cardiaci "brevi-lunghi-brevi". Tali fenomeni legati alla bradicardia sono più comuni nella forma acquisita della malattia. I segni clinici della forma congenita sono dovuti a mutazioni genetiche individuali. Sfortunatamente, la prima manifestazione clinica della malattia può essere la morte cardiaca improvvisa.
ECG. La durata dell'intervallo OT corretto è superiore a 460 ms e può raggiungere i 600 ms. Dalla natura dei cambiamenti nell'onda T, è possibile determinare una mutazione genetica specifica. Il normale intervallo WC in presenza della malattia nei familiari non esclude la possibilità di portatore. Il grado di prolungamento dell'intervallo OT varia, quindi in questi pazienti anche la varianza dell'intervallo OT risulta aumentata.
QT corretto normale - EXL/(intervallo RR) = 0,38-0,46 s (9-11 quadratini).
Sindrome del QT lungo: trattamento
Di solito gli episodi di aritmia come la "piroetta" sono di breve durata e scompaiono da soli. Gli episodi a lungo termine che causano disturbi emodinamici dovrebbero essere immediatamente eliminati con l'aiuto della cardioversione. In caso di convulsioni ricorrenti o dopo arresto cardiaco, viene somministrata per via endovenosa una soluzione di solfato di magnesio e poi flebo, quindi, se necessario, viene eseguita una stimolazione temporanea (frequenza 90-110). Come terapia preparatoria, si inizia un'infusione di isoprenalina prima della stimolazione.
Forma acquisita
Le cause dello sviluppo della sindrome dovrebbero essere identificate ed eliminate. È necessario interrompere l'assunzione di farmaci che causano l'allungamento dell'OT. Prima di ricevere i risultati degli esami del sangue, deve essere somministrato solfato di magnesio. È necessario determinare rapidamente il livello di potassio nel siero del sangue, la composizione del gas nel sangue. Con una diminuzione del livello di potassio inferiore a 4 mmol / l, è necessaria una correzione del suo livello al limite superiore della norma. Di solito non è necessario un trattamento a lungo termine, ma se la causa della condizione patologica è un blocco cardiaco irreversibile, è necessario un pacemaker permanente.
forma congenita
La maggior parte degli episodi sono provocati da un forte aumento dell'attività del sistema nervoso simpatico, pertanto il trattamento dovrebbe mirare a prevenire tali situazioni. I farmaci più preferiti sono i β-bloccanti. Il propranololo riduce il tasso di recidiva nei pazienti sintomatici. In assenza di effetto o intolleranza ai β-bloccanti, un’alternativa è la denervazione chirurgica del cuore.
La stimolazione cardiaca riduce i sintomi della bradicardia indotta dall'assunzione di β-bloccanti, nonché in situazioni in cui le pause nel lavoro del cuore provocano manifestazioni cliniche (LOT3). Nella forma congenita i pacemaker non sono mai considerati monoterapia. L’impianto di un defibrillatore dovrebbe essere preso in considerazione solo se esiste un rischio elevato di morte cardiaca improvvisa o se la prima manifestazione della malattia è stata una morte cardiaca improvvisa seguita da una rianimazione riuscita. L'installazione di un defibrillatore previene la morte cardiaca improvvisa, ma non previene il ripetersi di torsioni di punta. Possono verificarsi shock ripetitivi per brevi episodi
ridurre significativamente la qualità della vita dei pazienti. Un'attenta selezione dei pazienti, la nomina simultanea di β-bloccanti, la scelta della modalità di funzionamento dei defibrillatori aiutano a raggiungere il successo nel trattamento di tali pazienti.
Pazienti asintomatici
Lo screening tra i familiari del paziente consente di identificare individui con sindrome del QT lungo che non hanno mai avuto sintomi clinici. La maggior parte dei pazienti non muore a causa della sindrome del QT lungo, ma è a rischio di morte (il rischio nel corso della vita è del 13% se non trattati). È necessario valutare in ciascun caso il rapporto tra l'efficacia del trattamento permanente con il possibile sviluppo di effetti collaterali e il rischio di morte cardiaca improvvisa.
Determinare il rischio di sviluppare una morte improvvisa è un compito difficile, ma con una conoscenza accurata della natura dell'anomalia genetica diventa più semplice. Studi recenti hanno evidenziato la necessità di iniziare il trattamento in LOT1 con un allungamento dell'intervallo OT corretto superiore a 500 ms (sia per gli uomini che per le donne); con LQT2 - in tutti gli uomini e le donne con un aumento dell'intervallo QT superiore a 500 ms; a LQT3 - in tutti i pazienti. In ogni caso, è richiesto un approccio individuale.
La sindrome del QT lungo (LQT) è una patologia cardiaca congenita o acquisita, caratterizzata dall'allungamento dell'intervallo corrispondente, dalla presenza di sincopi ripetute e da un alto rischio di morte improvvisa dovuta allo sviluppo di aritmie maligne. La variante congenita della sindrome si manifesta in tutti i gruppi etnici con una frequenza compresa tra 1:2000 e 1:2500. Le femmine hanno maggiori probabilità di soffrirne. La prevalenza della sindrome acquisita varia da 2,5 a 4 casi per 1 milione di persone. Nel nostro articolo vedremo perché si verifica la LQT, quali sintomi provoca, perché è pericolosa e come trattarla.
La malattia è nota dalla fine del XIX secolo, quando fu descritta per la prima volta nella letteratura medica (1856, Meissner) l'osservazione di una ragazza con sordità congenita e frequenti svenimenti che si verificano con forte eccitazione. Successivamente fu rivelato il suo quadro elettrocardiografico (1953, Moller). Attualmente continua lo studio di questa sindrome e la ricerca di metodi efficaci per il suo trattamento.
Cause della sindrome congenita
La sindrome del QT lungo è caratterizzata da corrispondenti cambiamenti nell'elettrocardiogramma.La variante ereditaria della sindrome si basa su mutazioni nei geni che codificano le funzioni delle molecole proteiche dei canali ionici nel muscolo cardiaco. Attualmente, sono note più di 180 di tali mutazioni in 7 geni, che si trovano sul 3o, 7o, 11o e 21o cromosoma. Nella maggior parte dei casi, interrompono il lavoro dei canali del potassio e del sodio, meno spesso - dei canali del calcio e di specifiche proteine costruttive. Ciò porta ad un aumento della durata del potenziale d'azione nei cardiomiociti, dando inizio alla comparsa di tachicardia ventricolare del tipo "piroetta", che può trasformarsi in.
I processi di depolarizzazione e ripolarizzazione, che si verificano a seguito del movimento degli elettroliti nella cellula dallo spazio extracellulare e ritorno, riflettono l'intervallo QT sull'ECG, che in questa patologia è allungato.
Nella pratica clinica, ci sono 3 varianti principali della sindrome ereditaria:
- Romano-Ward (caratterizzato da un prolungamento isolato dell'intervallo QT, trasmesso da genitori con geni dominanti);
- Jervell-Lange-Nielsen (ereditato con modalità autosomica recessiva e associato a sordità congenita);
- variante autosomica dominante con manifestazioni extracardiache.
L'ultimo di essi può manifestarsi nella forma:
- Sindrome di Andersen-Tavila (il prolungamento dell'intervallo QT è combinato con un'onda U pronunciata, tachicardia ventricolare, anomalie nello sviluppo del sistema scheletrico, paralisi periodica iper o ipokaliemica);
- Sindrome di Timothy (sindattilia, anomalie cardiache congenite, vari disturbi della conduzione, rischio estremamente elevato di morte improvvisa).
Forma acquisita
In precedenza si credeva che l'insorgenza della sindrome LQT acquisita fosse associata a un malfunzionamento dei canali ionici, che non è causato da una mutazione, ma dall'influenza di fattori esterni o interni. Questa affermazione è vera, ma è stato dimostrato che un difetto genetico contribuisce allo sviluppo del processo patologico. Allo stesso tempo, la sindrome acquisita è difficile da distinguere dalla patologia congenita, poiché hanno molto in comune. Di solito, questa patologia passa inosservata per molto tempo e si manifesta in condizioni avverse, ad esempio sotto l'influenza di stress o sforzo fisico. I fattori che contribuiscono al prolungamento dell’intervallo QT includono:
- assumere farmaci (considereremo quali di seguito);
- disturbi elettrolitici (mancanza di potassio, sodio, magnesio);
- disturbi del ritmo cardiaco;
- malattie del sistema nervoso (lesioni, infezioni, tumori);
- cambiamenti nello stato ormonale (patologia della tiroide o delle ghiandole surrenali);
- alcolismo;
- fame, ecc.
Di particolare pericolo è l'esposizione di un organismo sensibile a diversi fattori di rischio.
Gruppi di farmaci che possono influenzare la lunghezza dell'intervallo QT
A causa del fatto che la sindrome LQT può essere causata dall'esposizione diretta ai farmaci e la loro cancellazione spesso porta alla normalizzazione di tutti gli indicatori, diamo uno sguardo più da vicino a quali farmaci possono modificare la lunghezza dell'intervallo QT:
- (amiodarone, novocainamide, sotalolo, propafenone, disopiramide);
- antibiotici (eritromicina, spiramicina, claritromicina, isoniazide);
- (ebastina, astemizolo);
- anestetici;
- antimicotici (fluconazolo, ketoconazolo);
- farmaci antitumorali;
- farmaci psicotropi (droperidolo, amitriptilina);
- (indapamide), ecc.
Non possono essere prescritti a soggetti che hanno già un prolungamento di tale intervallo. E con l'esordio tardivo della malattia, il loro ruolo come fattore provocatorio è necessariamente escluso.
Manifestazioni cliniche
 Questa malattia è caratterizzata da attacchi di improvvisa perdita di coscienza.
Questa malattia è caratterizzata da attacchi di improvvisa perdita di coscienza. Il quadro clinico della sindrome è caratterizzato dal polimorfismo dei sintomi. La loro gravità può variare da lievi vertigini alla perdita di coscienza e morte improvvisa. A volte quest'ultimo può fungere da primo segno della malattia. Le manifestazioni più tipiche di questa patologia sono:
- attacchi di perdita di coscienza;
- sordità congenita;
- casi di morte improvvisa in famiglia;
- cambiamenti nell'elettrocardiogramma (QT superiore a 450 ms, alternanza dell'onda T, tachicardia ventricolare del tipo "piroetta").
Con le varianti congenite della sindrome, è possibile rilevare altri sintomi caratteristici solo di essa.
Va notato che le condizioni sincopali in questa patologia hanno le loro caratteristiche:
- si verificano sullo sfondo dello stress, sotto l'influenza di forti stimoli sonori (sveglia, telefonata), attività fisica, sport (nuoto, immersioni), durante un brusco risveglio dal sonno notturno, nelle donne - dopo il parto;
- la presenza di sintomi che precedono la perdita di coscienza (grave debolezza, ronzio nelle orecchie, oscuramento degli occhi, sensazione, pesantezza dietro lo sterno);
- rapido recupero della coscienza con esito favorevole;
- mancanza di amnesia e cambiamenti di personalità (come nell'epilessia).
A volte la perdita di coscienza può essere accompagnata da convulsioni e minzione involontaria. In tali casi viene effettuata la diagnosi differenziale con crisi epilettiche.
Il corso del processo patologico in ciascun paziente può presentare alcune differenze. Dipende sia dal genotipo che dalle condizioni di vita. Le seguenti opzioni sono considerate le più comuni:
- sincope che si verifica sullo sfondo del prolungamento dell'intervallo QT;
- allungamento isolato di questo intervallo;
- sincope in assenza di alterazioni dell'ECG;
- completa assenza di sintomi (alto rischio senza manifestazioni fenotipiche della malattia).
Il decorso più sfavorevole è complicato dallo sviluppo della fibrillazione ventricolare e dell'arresto cardiaco.
Con le varianti congenite della malattia, lo svenimento si verifica durante l'infanzia (5-15 anni). Inoltre, la loro presenza nei bambini in età prescolare è un segno prognosticamente sfavorevole. E il parossismo della tachicardia ventricolare, che ha richiesto cure di emergenza, aumenta di 10 volte la probabilità di un secondo arresto cardiaco nel prossimo futuro.
I pazienti con sindrome del QT lungo asintomatica possono non essere consapevoli della loro diagnosi e avere un'aspettativa di vita normale, ma trasmettono la mutazione ai loro figli. Questo flusso è osservato molto spesso.
Principi diagnostici
La diagnosi della sindrome si basa sui reperti clinici e sui risultati dell'elettrocardiografia. Il monitoraggio Holter fornisce ulteriori informazioni al medico.
Considerando che non è sempre facile fare una diagnosi, sono stati sviluppati criteri diagnostici maggiori e minori. Questi ultimi includono:
- perdita dell'udito dalla nascita
- variabilità dell'onda T in diverse derivazioni (sull'elettrocardiogramma);
- violazione dei processi di ripolarizzazione del miocardio dei ventricoli;
- frequenza cardiaca bassa.
I criteri principali includono:
- prolungamento dell'intervallo QT corretto oltre 450 ms a riposo;
- episodi di perdita di coscienza;
- casi di malattia in famiglia.
La diagnosi è considerata affidabile in presenza di due criteri maggiori o di uno maggiore e due minori.
Trattamento
 A causa dell'inefficacia di altre misure terapeutiche, il paziente necessita dell'impianto di un defibrillatore cardioverter.
A causa dell'inefficacia di altre misure terapeutiche, il paziente necessita dell'impianto di un defibrillatore cardioverter. La direzione principale del trattamento di tali pazienti è la prevenzione delle aritmie maligne e dell'arresto cardiaco.
Tutti gli individui con un intervallo QT lungo dovrebbero evitare:
- situazioni stressanti;
- Fare sport;
- sforzo fisico intenso;
- assumere farmaci che aumentano la durata di questo intervallo.
Tra i farmaci per questa sindrome, di solito vengono prescritti i seguenti:
- β-bloccanti;
- preparati di magnesio e potassio;
- mexiletina o flecainide (basse dosi).
Con l'inefficacia della terapia conservativa, si ricorre alla denervazione simpatica o all'impianto di un defibrillatore cardioverter. Quest'ultimo è particolarmente importante nei pazienti ad alto rischio di morte cardiaca improvvisa e sottoposti a rianimazione.
Riso. 2-12. Misura dell'intervallo Q-T. R-R è l'intervallo tra due complessi QRS consecutivi.
Valore dell'intervallo Q-T
Innanzitutto questo intervallo riflette il ritorno dei ventricoli dallo stato di eccitazione allo stato di riposo (ventricoli). Valore dell'intervallo normale Q-Tdipende dalla frequenza cardiaca. Con un aumento della frequenza del ritmo [accorciando l'intervallo R-R(intervallo tra successivi)] è caratterizzato da un accorciamento dell'intervallo Q-T, quando il ritmo rallenta (allungando l'intervallo R-R) - allungamento dell'intervallo Q-T.
Regole per misurare l'intervallo Q-T
Quando l'intervallo Q-T allungato, misurare spesso difficile a causa della fusione poco appariscente della parte finale con . Di conseguenza, è possibile misurare l'intervallo QU, ma no Q-T.
Nella tabella. 2-1 sono indicati i valori approssimativi del limite superiore dell'intervallo normale Q-T per frequenze cardiache diverse. Sfortunatamente, non esiste un modo più semplice per determinare il valore Q-T normale non esiste. Viene proposto un altro indicatore: intervallo corretto Q-T a seconda della frequenza del ritmo. Intervallo corretto Q-T (Q-T K) si ottiene dividendo la durata dell'intervallo effettivo Q-T alla radice quadrata dell'intervallo R-R(entrambi i valori sono in secondi):
QT C = (QT) ÷ (√RR)
L'intervallo normale Q-T non supera 0,44 s. Per calcolare l'intervallo Q-T a seconda della frequenza del ritmo sono state proposte altre formule, ma non tutte sono universali. Alcuni autori chiamano limite superiore Q-T y uomini 0,43 s, donne - 0,45 s.

Cambiamenti nella lunghezza dell'intervallo Q-T
Allungamento patologico dell'intervallo Q-T molti fattori possono contribuire (Figura 2-13).

Riso. 2-13. Prolungamento dell'intervallo Q-T in un paziente che assume chinidina. L'effettivo intervallo Q-T (0,6 s) è significativamente prolungato per questa frequenza (65 bpm); anche l'intervallo QT corretto (normalmente inferiore a 0,44 s) risulta allungato (0,63 s); il rallentamento della ripolarizzazione ventricolare predispone allo sviluppo di tachicardia ventricolare potenzialmente letale del tipo "piroetta"; il calcolo dell'intervallo Q-T in questo caso viene eseguito come segue: QTC = (QT) ? (?RR) = 0,60? ?0,92 = 0,63
Ad esempio, alcuni (amiodarone, disopiramide, dofetilid, ibutilide, procainamide, chinidina, sotalolo), antidepressivi triciclici (fenotiazine, pentamidina, ecc.) possono aumentarne la durata. Anche i disturbi elettrolitici (diminuzione dei livelli di potassio, magnesio o calcio) sono considerati un'importante causa di prolungamento dell'intervallo. Q-T.
Ipotermia contribuisce inoltre al suo allungamento rallentando la ripolarizzazione delle cellule del miocardio. Altri motivi per l'allungamento dell'intervallo Q-T-, infarto del miocardio (soprattutto in fase acuta) ed emorragie subaracnoidee. Aumentare la durata dell'intervallo Q-T predispone allo sviluppo di aritmie ventricolari potenzialmente letali [(TV) del tipo "piroetta" (torsades de pointes)]. Diagnosi differenziale delle condizioni con un intervallo esteso Q-T descritto nel cap. 24.
- In contatto con 0
- Google Plus 0
- OK 0
- Facebook 0






