Цель занятия : Изучить общую клиническую характеристику аутосомных и гоносомных болезней. Определить основные показания к назначению цитогенетического обследования у детей и взрослых. Научиться читать кариотип.
План занятия :
1. Определение термина «Хромосомное заболевание».
2. Нормальный кариотип, его характеристики. Гомологичные хромосомы.
3. Этиология хромосомных болезней: геномные и хромосомные мутации.
4. Общие правила записи хромосомных аномалий и чтения кариотипа.
5. Общая клиническая характеристика аутосомных болезней. Наиболее часто встречающиеся заболевания.
6. Общая клиническая характеристика гоносомных болезней.
7. Хромосомные аномалии у клинически здоровых лиц.
8. Показания к назначению цитогенетического обследования.
Основные термины:
Кариотип – число и совокупность морфологических особенностей полного хромосомного набора человека.
Идиограмма - Схематичное изображение дифференциальной исчерченности хромосомы, позволяющее определить правильность ее структуры.
Гомологичные хромосомы - парные хромосомы, которые содержат один и тот же набор генов, сходны по морфологическому строению, конъюгируют в мейозе и могут обмениваться участками в процессе кроссинговера. В отличие от сестринских хроматид, синтезированных на основе единственной молекулы ДНК, гомологичные хромосомы не идентичны по нуклеотидному составу и могут нести различные аллельные формы одного и того же гена.
Геномные мутации - числовые аномалии отклонения от нормального числа хромосом (46).
Полиплоидия - изменения числа хромосом, кратные гаплоидному набору:
Анеуплоидия – изменения числа хромосом, не кратные гаплоидному набору, как правило, в пределах одной пары гомологов.
Структурные перестройки хромосом (хромосомные мутации) – нарушения структуры хромосом, приводящие или не приводящие к хромосомному дисбалансу.
Мозаицизм – одновременное наличие в организме нескольких клонов, имеющих разный кариотип.
Трисомия - наличие третьей гомологичной хромосомы.
Моносомия - отсутствие одного из гомологов.
Полисомия - увеличение числа хромосом в пределах одной пары гомологов
Аутосомы – все хромосомы организма, за исключением половых.
Гоносомы – половые хромосомы.
Основные классификации:
- Геномные мутации (числовые аномалии)
- Полиплоидии у человека встречаются триплоидия 69 хромосом (3n) - приводят к прерыванию беременности или гибели ребенка вскоре после рождения, тетраплоидия –92 хромосомы (4n) –летальны, приводят к прерыванию беременности на ранних сроках.
- Анеуплоидии
- Моносомии – отсутствие одной из пары гомологичных хромосом. Единственная известная форма 45,Х.
- Трисомии – наличие третьей гомологичной хромосомы. Причина наиболее распространенных хромосомных болезней. Синдром Дауна – трисомия 21, синдром Патау – трисомия 13, синдром Эдвардса – трисомия 18.
- Полисомии (тетрасомия , пентасомия ) – наличие нескольких (четырех, пяти) гомологичных хромосом. У живорожденных описано только для половых хромосом. Например, 48,ХХХХ.
Для обозначения числовых аномалий аутосом при записи кариотипа используют следующий подход – 1) цифра, указывающая общее число хромосом, 2) формула набора половых хромосом. 3) знаки (+) или (-), за которыми следует число утраченных или лишних хромосом. Мальчик с синдромом Дауна 47, XY, +21
- Хромосомные мутации (структурные перестройки)
- Сбалансированны е
- Инверсии (i nv) поворот участка хромосомы на 180 градусов без утраты генетического материала.
- Транслокации (t)– обмен генетическим материалом между хромосомами
- Робертсоновские транслокации(rob) – объединение двух акроцентрических хромосом в единую морфологическую структуру
- Несбалансированные
- Делеции (del)– утрата части генетического материала
- Дупликации (dup) - изменение хромосомы, при котором один из её участков линейно представлен два или более раз.
- Изохромосомы(i) – хромосомы, состоящие из двух коротких или двух длинных плеч с одновременной утратой материала другого плеча.
Сбалансированные перестройки не приводят к возникновению хромосомных заболеваний у носителей, но могут явиться причиной бесплодия, привычного невынашивания беременности или рождения детей с хромосомными болезнями. Несбалансированные перестройки являются причиной хромосомных болезней.
Запись структурной перестройки в кариотипе всегда начинается с одно- или трёхбуквенной аббревиатуры, затем чаще всего перечисляются в скобках вовлечённые хромосомы и точки разрыва. Например: 46,ХХ,ins(7;2)(p13;q22q32). Согласно международной номенклатуре, аббревиатуры структурных перестроек следующие: inv – инверсия, t – реципрокная транслокация, ins – нереципрокная транслокация или инсерция, rob – робертсоновская транслокация, del – делеция, r – кольцевая хромосома, dup – дупликация, i – изохромосома.
Порядок записи структурных аномалий. После числа хромосом и набора половых хромосом название перестройки с использованием стандартных символов, затем в круглых скобках хромосомы, вовлеченные в перестройку. Сначала половые, потом аутосомы. Разделяются (;) В следующих круглых скобках точки разрыва, также разделяются (;).
Классификация хромосомных болезней. Клинически подразделяют аутосомные болезни и нарушения в системе половых хромосом (гоносомные заболевания).
Аутосомные болезни – полные трисомии, мозаичные трисомии. Структурные перестройки, возникшие de novo или как результат сбалансированных родительских перестроек, клинически обозначают частичные трисомии или частичные моносомии .
Гоносомные болезни моносомия, трисомии, полисомии, структурные перестройки.
Основные схемы и рисунки:
Кариограмма нормального хромосомного набора. Слева от каждой хромосомы – ее идиограмма.
Основные описания:
Несмотря на то, что такие хромосомные болезни, как синдром Дауна, синдром Эдвардса, некоторые наиболее часто встречающиеся синдромы частичных трисомий и моносомий имеют характерную клиническую картину, окончательный диагноз ставится только после кариотипирования. Кроме того, многие случаи несбалансированного кариотипа с незначительным хромосомным дисбалансом, не дают четкой картины хромосомного синдрома. Поэтому, прежде всего, необходимо представлять, в каких случаях требуется направление на кариотипирование.
Аутосомное заболевание следует подозревать в случае сочетания трёх ситуаций у пациента:
а) Множественные врождённые пороки развития, т.е. наличие как минимум двух врождённых пороков развития, патогенетически не связанных друг с другом и затрагивающих как минимум две системы органов.
б) Множественные внешние аномалии и микроаномалии развития
в) Задержка психомоторного развития ребёнка или нарушения интеллекта ребенка.
Сочетание этих условий встречается во всех случаях хромосомных болезней, наиболее частыми из которых являются:
Синдром Дауна. Существуют три цитогенетические формы синдрома Дауна: регулярная трисомия (93% всех случаев), транслокационная (5%) и мозаичная (2%) формы. Основными клиническими признаками синдрома Дауна являются умственная отсталость, мышечная гипотония, эпикант и монголоидный разрез глазных щелей, косоглазие, толстые губы, утолщенный язык с бороздами (так называемый “географический или складчатый язык”), плоская спинка носа, узкое небо, деформированные ушные раковины, избыток кожи на шее, поперечная линия ладони (так называемая “обезьянья складка”), клинодактилия мизинцев. Среди аномалий внутренних органов отмечают пороки сердца (дефекты перегородок в сочетании с аномалиями крупных сосудов), желудочно-кишечного тракта, мочевой системы, мозга. Дети с синдромом Дауна страдают глубокой умственной отсталостью в степени имбецильности. В разной степени страдают отдельные виды психической деятельности.
Синдром Патау. Синдром трисомии хромосомы 13. В основном встречается как регулярная трисомия, структурные перестройки, как правило, в виде робертсоновских транслокаций, описаны лишь в небольшом количестве случаев. Дети с синдромом Патау рождаются с пренатальной гипоплазией (масса тела до 2500г). Основными признаками синдрома Патау является расщелина верхней губы и неба, дефекты скальпа. Центральная нервная система поражена во всех случаях синдрома, часты аплазия и гипоплазия червя мозжечка. При данном синдроме характерны следующие проявления: микроцефалия, тригоноцефалия, низкий скошенный лоб, широкий нос с запавшей переносицей, узкие глазные щели, гипертелоризм, микрофтальмия (реже анофтальмия), колобома радужки, полидактилия кистей и стоп, флексорное положение кистей, “стопа качалка”, крипторхизм, гипоспадия, гипоплазия полового члена, удвоение матки и влагалища. Множественные аномалии внутренних органов, сердечно-сосудистой системы (дефекты межжелудочковой и межпредсердной перегородки, пороки крупных сосудов). Дети с синдромом Патау в основном умирают в первые дни и месяцы жизни от тяжелых пороков развития, несовместимых с жизнью. В единичных случаях продолжительности жизни до 2-3х лет наблюдается глубокая идиотия.
Синдром Эдвардса , трисомия хромосомы 18. Дети рождаются с пренатальной гипоплазией (масса тела при рождении не более 2200). Фенотипические проявления при синдроме Эдвардса характерны и многообразны. К ним относятся долихоцефалия, микрофтальмия, низко расположенные деформированные ушные раковины, высокое небо, расщелина неба, микрогения, микростомия, гипертрофия клитора, гипоспадия, крипторхизм, аномалии конечностей (флексорное положение кистей, короткий и широкий большой палец стопы, “стопа-качалка”, кожная синдактилия стоп, косолапость). Из пороков внутренних органов наблюдают аномалии сердца (дефекты межпредсердной и межжелудочковой перегородки), пищеварения, мочевой системы, ЦНС (гипоплазия мозжечка и мозолистого тела). Нарушения развития головного мозга наблюдают во всех случаях. В большинстве случаев дети умирают на первом году жизни. Редкие случаи продолжительности жизни до 3-5 лет связаны, как правило, с мозаичными вариантами.
Наиболее частым из синдромов, связанных с делециями аутосомявляется синдром «крика кошки» - моносомия короткого плеча хромосомы 5 (5р-). Кариотип 46,ХХ,del(5p), 46,XY,del(5p). Основные признаки - специфический, напоминающий кошачье мяуканье крик, связанный с изменениями гортани, который, как правило, исчезает к году, микроцефалия, лунообразное лицо, антимонголоидный разрез глаз, эпикант, косоглазие, гипертелоризм, маленькая нижняя челюсть, низко расположенные ушные раковины, клинодактилия, синдактилия.
Другими относительно частыми делеционными синдромами являются синдром Вольфа-Хиршхорна _ del(4p), синдром Орбели - del 13q, синдромы del 18(q) и del 18(p). Из синдромов частичных трисомий наиболее распространенным является синдром Реторе. Все они характеризуются резкой задержкой физического и психомоторного развития, глубокой умственной отсталостью. Во всех случаях наблюдаются микроцефалия различной степени, лицевые дизморфизмы, аномалии носа и глаз, деформированные, низко расположенные ушные раковины. Из внутренних органов чаще поражаются сердце, почки и органы ЖКТ. Могут наблюдаться аномалии конечностей. Прогноз для жизни неблагоприятный, большинство больных погибают в раннем детском возрасте. Сочетание этих аномалий является основанием для того, чтобы предполжить наличие хромосомного дисбаланса,. показанием для направления на консультацию к генетику и дальнейшего цитогенетического обследования.
Гоносомные болезни , в отличие от аутосомных, редко приводят к тяжелым отклонениям в состоянии здоровья и интеллекта носителя таких аномалий
МВПР нет
Множественных внешних аномалий нет
Интеллект нормальный, слегка снижен или изменён качественно
Зачастую страдают наружные и внутренние гениталии, рост и телосложение
Продолжительность жизни в значимой степени не снижена
Для гоносомных заболеваний характерна широкая вариабельность клинических проявлений от наличия классического фенотипа, позволяющего осуществлять «портретную диагностику» и сразу заподозрить аномалии кариотипа, до относительно мягких проявлений, когда направление к генетику проводят лишь после длительного безрезультатного лечения репродуктивных проблем. Однако в большинстве случаев заподозрить гоносомное заболевание можно по следующим признакам: а) девиации роста. б) изменение телосложения, задержка полового развития, аномалии наружных и внутренних гениталий. в) у взрослых признаками хромосомных аномалий могут служить бесплодие, нарушения менструального цикла у женщин, азооспермия у мужчин.
Синдром Клайнфельтера . Вариант, обусловленный дисомией Х хромосомы. Кариотип 47,ХХY. Мужской фенотип. Рост различный. Евнухоидное телосложение, горизонтальный рост волос на лобке, гинекомастия. Наружные половые органы сформированы по мужскому типу, но гипоплазированы, резко уменьшен размер яичек и, именно микроорхидизм считается одним из важных клинических критериев в диагностике синдрома. У больных с кариотипом 47,ХХY абсолютное бесплодие, вызванное гиалинизацией семенных канальцев и азооспермией. Больные с мозаичными формами могут быть фертильны. Для большинства больных характерна мужская психосексуальная ориентация. Половая функция сохранна. Интеллект колеблется от практически не измененного (в большинстве случаев) до олигофрении. Около 25% случаев заболевания диагностируется только у взрослых при обращении к генетику в связи с бесплодием, значительная часть больных остается недиагностированной. Полисомные формы синдрома Клайнфельтера, когда в кариотипе появляются две или три дополнительных Х хромосомы имеют более тяжелое течение и сопровождаются умственной отсталостью.
Синдром Шерешевского-Тернера .. Кариотип 45,Х, либо структурно измененная Х-хромосома. Из структурных нарушений наиболее распространены цитогенетические варианты: 46,Х,i(Х)(q10), 46,Х,del(X), 46,X,r(X), почти в половине случаев наблюдаются мозаичные формы. Женский фенотип. Низкий рост, задержка полового развития. Гипоплазия/аплазия матки и яичников. В большинстве случаев – аменорея и бесплодие. Внешние аномалии.: гипертелоризм сосков и втянутые соски, вальгусная деформация локтевых суставов, крыловидные складки шеи, изменение пальцевой дуги на ладонях и стопах, щитообразная грудная клетка, низкий рост волос на затылке и пр. В 10 – 20% случаев наблюдается коарктация аорты. Внешние аномалии обычно встречаются в случаях полной моносомии Х, при структурных аномалиях они менее выражены, либо отсутствуют совсем. Интеллект нормальный. Продолжительность жизни слегка снижена. При внешних аномалиях диагноз может ставиться уже при рождении, при этом у новорожденных часто наблюдается лимфатический отек кистей и стоп. При отсутствии выраженных внешних аномалий причиной обращения у девочек дошкольниц может служить выраженная задержка роста, в более старшем возрасте – задержка полового развития. Около 10 % случаев диагностируется только у взрослых, как правило, при обследовании по поводу бесплодия.
Синдром XYY . Частота 1:1000. Кариотип 47,XYY. Мужской фенотип. Как правило, фертильны, хотя нередко встречаются среди пациентов с бесплодием. Вследствие отсутствия выраженных клинических проявлений диагностируется редко.
Синдром трисомии Х . Частота 1:1000. Кариотип 47,ХХХ. Женский фенотип. Нормальный рост. Правильное телосложение и черты лица. Различные нарушения менструального цикла, часто вторичная аменорея, преждевременное истощение яичников. Иногда наблюдается незначительное снижение интеллекта. Больные, как правило, фертильны, но в старшем репродуктивном возрасте возможно бесплодие, связанное с ранним угасанием функции яичников. Диагностируется редко.
Среди перинатально погибших плодов частота хромосомных аномалий составляет 6%.
Наиболее тяжелые формы по дисбалансу хромосомного набора встречаются у ранних абортусов. Это полиплоидии (25%), полные трисомии по аутосомам (50%). Трисомии по некоторым аутосомам (1; 5; 6; 11; 19) встречаются крайне редко даже у элиминированных эмбрионов и плодов, что свидетельствует о большой морфогенетической значимости генов в этих аутосомах. Данные аномалии прерывают развитие в доимплантационном периоде или нарушают гаметогенез.
Высокая морфогенетическая значимость аутосом еще более отчетлива при полных аутосомных моносомиях. Последние редко обнаруживаются даже в материале ранних спонтанных абортов из-за летального эффекта такого дисбаланса.
Врожденные пороки развития
Если хромосомная аномалия не дает летального эффекта на ранних стадиях развития, то ее последствия проявляются в виде врожденных пороков развития. Практически все хромосомные аномалии (кроме сбалансированных) приводят к врожденным порокам
развития, сочетания которых известны как нозологические формы хромосомных болезней и синдромов (синдромы Дауна, Вольфа- Хиршхорна, кошачьего крика и т.д.).
С эффектами, вызываемыми однородительскими дисомами, подробнее можно ознакомится на компакт-диске в статье С.А. Назаренко «Наследственные болезни, детерминированное однородительскими дисомами, и их молекулярная диагностика».
Эффекты хромосомных аномалий в соматических клетках
Роль хромосомных и геномных мутаций не ограничивается их влиянием на развитие патологических процессов в ранних периодах онтогенеза (незачатие, спонтанный аборт, мертворождение, хромосомная болезнь). Их эффекты прослеживаются в течение всей жизни.
Хромосомные аномалии, возникающие в соматических клетках в постнатальном периоде, могут вызывать различные последствия: остаться нейтральными для клетки, обусловить гибель клетки, активировать деление клетки, изменить функцию. Хромосомные аномалии возникают в соматических клетках постоянно с невысокой частотой (около 2%). В норме такие клетки элиминируются иммунной системой, если они проявляют себя чужеродно. Однако в некоторых случаях (активация онкогенов при транслокациях, делециях) хромосомные аномалии становятся причиной злокачественного роста. Например, транслокация между хромосомами 9 и 22 вызывает миелолейкоз. Облучение и химические мутагены индуцируют хромосомные аберрации. Такие клетки гибнут, что наряду с действием других факторов способствует развитию лучевой болезни, аплазии костного мозга. Имеются экспериментальные доказательства накопления клеток с хромосомными аберрациями в процессе старения.
Причинами этой группы заболевания являются изменения числа аутосом (по типу моносомии или трисомии) или различные хромосомные мутации аутосом. Наиболее часто встречаются нарушения по 8, 9, 13 (синдром Патау), 18 (синдром Эдвардса), 21 (синдром Дауна) хромосомам.
Для диагностики болезни необходимо определить:
Тип мутации (геномная или хромосомная)
Вовлеченную в процесс хромосому
Форму (полная или мозаичная)
Вид болезни (спорадический случай или наследственная форма)
Мутации аутосом вызывают нарушение общего генетического баланса клеток, поэтому патологические эффекты проявляются на всех этапах онтогенеза и в различных органах. Главные эффекты мутаций проявляются в двух вариантах: летальности и врожденных пороках развития. Летальный эффект – один из главных факторов внутриутробной гибели плода. Родившиеся живыми младенцы имеют многочисленные нарушения развития. При различных хромосомных аномалиях наблюдаются сходные клинические проявления: низкий вес при рождении, задержка физического и умственного развития, нарушения формирования лицевого и мозгового черепа (деформация ушных раковин, микроцефалия, мозговые грыжи),
пороки развития внутренних органов. Если при рождении у младенца выявляются несколько пороков развития, то в первую очередь следует подумать о наличии у него хромосомного заболевания.
Синдром Дауна. Встречается с частотой 1:700-1:800 родов, при этом отсутствует зависимость от пола ребенка. Риск рождения больного ребенка увеличивается с возрастом матери и в меньшей степени зависит от возраста отца.
Цитогенетические варианты болезни разнообразны: 95% - полная трисомия (при этом «материнский вклад» - 80%), транслокационная форма (21 хромосома + 15 хромосома) – данная форма болезни может наследоваться от родителей.
Больные дети разных рас удивительно похожи друг на друга. Больные имеют характерный внешний вид: монголоидный разрез глаз, круглое уплощенное лицо, плоская спинка носа, крупный (обычно высунутый) язык, деформированные уши. Характерны дерматоглифические особенности: «обезьянья» складка на ладони, две кожные складки на мизинце. У больных выявляются пороки внутренних органов, чаще сердечно-сосудистой системы, органов пищеварения, почек.
У больных нарушается физическое и умственное развитие: рост взрослых больных на 20 см ниже среднего, умственная отсталость (олигофрения) разных степеней. Больные ласковые, послушные, могут осваивать несложные практические навыки. Однако снижение мышечного тонуса в сочетании с разболтанностью суставов не позволяет выполнять тонкие координированные движения. Средняя продолжительность жизни составляет 30-35 лет.
Синдром «кошачьего крика» обусловлен выпадением (делецией) участка короткого плеча 5 хромосомы (кариотип: 46, 5 p-). У детей наблюдается необычный плач, напоминающий требовательное кошачье мяуканье. Клиническая картина значительно варьирует у разных больных. Характерный крик обусловлен изменением гортани (сужение, мягкость хрящей, уменьшение надгортанника). Практически у всех больных отмечаются изменения строения мозгового и лицевого черепа: лунообразное лицо, микроцефалия. Ушные раковины деформированы и низко расположены. Встречаются пороки сердца и костно-мышечной системы: синдактилия, косолапость. Большинство больных умирает в возрасте до 1 года.
В случае гетероплоидии особенно тяжелы моносомии. Моносомии по аутосомам заканчиваются летально еще в первые дни эмбрионального развития или приводят к гибели зародыша на более поздних стадиях (спонтанные аборты). Полные трисомии описаны у человека по большому количеству хромосом: 8, 9, 13, 14, 18, 21, X, Y. Наиболее изученными синдромами, в основе которых лежат нарушения в системе аутосом (геномные мутации, хромосомные мутации) являются трисомии 21, 13, 18, транслокационная форма Дауна, синдром «кошачьего крика», в системе половых хромосом трисомии XXY, XXX, XYY и моносомия XO.
болезнь дауна
(трисомия 21; 47,XX(XY)+21 )
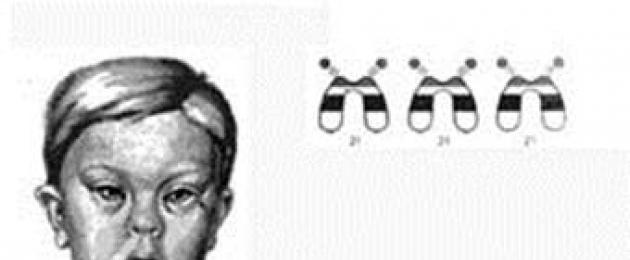
Диагностика болезни Дауна уже у новорожденного не вызывает затруднений (рис. 5). При болезни Дауна встречается от 9 до 29 соматических аномалий. Чаще при этом синдроме имеются:
· Брахицефальный череп со сглаженным затылком и уплощенным лицом, эпикант;
· Пятна Брушфильда (светлые пятна на радужке);
· Маленькие недоразвитые ушные раковины;
· Увеличенный «складчатый» язык;
· Широкие кисти с короткими пальцами и укороченными искривленными пятыми пальцами (клинодактилия);
· Поперечная борозда на одной или обеих ладонях («обезьянья складка»);
· Расширенные промежутки между 1 и 2-м пальцами стоп.
 |
| Рис.5. Симптомы трисомии 21 |
Интеллектуальный дефект больных углубляется с возрастом. Известно, что примерно у 60% детей с болезнью Дауна имеются разные формы глазной патологии а у 70% обнаруживают тугоухость.
Большое внимание в последние годы уделяется изучению патогенеза синдрома Дауна. Сегодня предложена объединенная генетическая гипотеза синдрома Дауна и болезни Альцгеймера. В статусе таких больных выявляется преждевременное старение, преобладание дегенеративных сосудистых нарушений, сахарный диабет, катаракта͵ липофусциноз, амилоидоз, избирательное повреждение холинергических нейронов в базальных ганглиях, склонность к злокачественным новообразованиям, специфические нарушения слуха и другие признаки, а главное – характерные нарушения интеллекта͵ напоминающие таковые при старческой болезни Альцгеймера.
Использование цитогенетичеcких методов исследования показало, что примерно 80% всех случаев простой трисомии 21 имеет материнское происхождение и около 20% – отцовское. При этом лишь 20% всех случаев «материнского» синдрома Дауна обусловлено нерасхождением хромосом 21-ой пары во втором делении мейоза, а остальные – ошибками первого деления мейоза.
Болезнь дауна транслокационной формы
Транслокационные формы синдрома Дауна наблюдаются в 3-4% случаев. Число хромосом в данном варианте болезни нормальное – 46, так как дополнительная хромосома 21 транслоцирована на аутосомы 13, 14, 15 и 22 (рис.6)
В случае гетероплоидии особенно тяжелы моносомии. Моносомии по аутосомам заканчиваются летально еще в первые дни эмбрионального развития или приводят к гибели зародыша на более поздних стадиях (спонтанные аборты). Полные трисомии описаны у человека по большому количеству хромосом: 8, 9, 13, 14, 18, 21, X, Y. Наиболее изученными синдромами, в основе которых лежат нарушения в системе аутосом (геномные мутации, хромосомные мутации) являются трисомии 21, 13, 18, транслокационная форма Дауна, синдром «кошачьего крика», в системе половых хромосом трисомии XXY, XXX, XYY и моносомия XO.
болезнь дауна
(трисомия 21; 47,XX(XY)+21 )
Диагностика болезни Дауна уже у новорожденного не вызывает затруднений (рис. 5). При болезни Дауна встречается от 9 до 29 соматических аномалий. Чаще при этом синдроме имеются:
· Брахицефальный череп со сглаженным затылком и уплощенным лицом, эпикант;
· Пятна Брушфильда (светлые пятна на радужке);
· Маленькие недоразвитые ушные раковины;
· Увеличенный «складчатый» язык;
· Широкие кисти с короткими пальцами и укороченными искривленными пятыми пальцами (клинодактилия);
· Поперечная борозда на одной или обеих ладонях («обезьянья складка»);
· Расширенные промежутки между 1 и 2-м пальцами стоп.
| Рис.5. Симптомы трисомии 21 |
Интеллектуальный дефект больных углубляется с возрастом. Известно, что примерно у 60% детей с болезнью Дауна имеются разные формы глазной патологии а у 70% обнаруживают тугоухость.
Большое внимание в последние годы уделяется изучению патогенеза синдрома Дауна. В настоящее время предложена объединенная генетическая гипотеза синдрома Дауна и болезни Альцгеймера. В статусе таких больных выявляется преждевременное старение, преобладание дегенеративных сосудистых нарушений, сахарный диабет, катаракта, липофусциноз, амилоидоз, избирательное повреждение холинергических нейронов в базальных ганглиях, склонность к злокачественным новообразованиям, специфические нарушения слуха и другие признаки, а главное – характерные нарушения интеллекта, напоминающие таковые при старческой болезни Альцгеймера.
Использование цитогенетичеcких методов исследования показало, что примерно 80% всех случаев простой трисомии 21 имеет материнское происхождение и около 20% – отцовское. При этом лишь 20% всех случаев «материнского» синдрома Дауна обусловлено нерасхождением хромосом 21-ой пары во втором делении мейоза, а остальные – ошибками первого деления мейоза.
Болезнь дауна транслокационной формы
Транслокационные формы синдрома Дауна наблюдаются в 3-4% случаев. Число хромосом в данном варианте болезни нормальное – 46, так как дополнительная хромосома 21 транслоцирована на аутосомы 13, 14, 15 и 22 (рис.6)
При транслокационном варианте синдрома Дауна один из фенотипически здоровых родителей может быть носителем сбалансированной транслокации. В кариотипе этих родителей имеется по 45 хромосом. Одна хромосома состоит как бы из двух частей и содержит генетический материал недостающей хромосомы (рис.8), поэтому при общем числе хромосом, равном 45, нет утери генетического материала, а перестройка сбалансирована. Примерно в 1/3 всех случаев транслокационный вариант синдрома Дауна имеет наследственный характер. Выявление у кого-либо из родителей сбалансированной транслокации определяет необходимость пренатальной диагностики.
Синдром эдвардса
(трисомия 18; 47, XX(XY)+18 )
При кариологичеком обследовании больных выявляется лишняя хромосома из группы Е (хромосома 18), (рис. 7).
 |
| Рис. 7. Симптомы трисомии 18 |
Фенотипические проявления синдрома Эдвардса довольно характерны:
· Долихоцефальный череп, сдавленный с боков, с низким лбом и широким выступающим затылком;
· Глазные щели узкие; эпикант;
· Нижняя челюсть маленькая, скошена назад (микроретрогнатия);
· Рот маленький, треугольной формы с короткой верхней губой;
· Шея короткая, с крыловидной складкой.
Аномалии опорно-двигательного аппарата:
- Кисти и пальцы короткие, пятые пальцы искривлены, пальцы сжаты в кулак, второй и пятый пальцы расположены сверху и прикрывают прижатые к ладони второй и четвертый пальцы;
- Первый палец стопы короткий и широкий, синдактилия второго и третьего пальцев;
- Форма стопы в виде «качалки».
Почти 95% больных имеют пороки сердца, крупных сосудов, мочеполовой системы, аномалии органов пищеварения. Прогноз для жизни неблагоприятный.
Синдром патау
(трисомия 13; 47, XX(XY)+13 )
При кариологическом анализе соматических клеток больных выявляется лишняя хромосома из группы D (хромосома 13) (рис. 8).
Клиническая картина типична:
· Микроцефальный череп с низким скошенным лбом и вдавленными височными областями;
· Глазные щели узкие, расположены горизонтально, растояние между ними уменьшено (гипотелоризм), почти всегда встречается глазная патология;
· Ушные раковины расположены низко, маленькие мочки прижаты к голове, завитки неправильной формы;
· Череп с углублениями в теменно-затылочной области, растояние между теменными буграми увеличено;
· Демонстративным признаком синдрома Патау являются «заячья губа» и «волчья пасть». Расщелины могут быть как двусторонними, так и односторонними. Почти всегда расщепление верхней губы сопровождается расщелиной неба.
 |
| Рис. 8. Симптомы трисомии 13 |
Аномалии костно-мышечной системы:
· Полидактилия на верхних и нижних конечностях;
· Второй и четвертый пальцы согнуты, приведены к ладони и перекрыты первым и пятым пальцами;
· Выявляются дефекты развития практически всех систем и органов;
· Мозг часто не разделен на полушария, гипоплазия лобных долей, мозжечка.
У 50% больных выявляются пороки развития мочевыводящих путей: кистозная почка, гидронефроз, дисплазия почек, у 50% девочек находят удвоение влагалища и двурогую матку с гипоплазией яичников. Прогноз для жизни неблагоприятный.
Синдром «кошачьего крика»
(синдром 5р–)
Наиболее частый из всех синдромов делеции аутосом – синдром делеции короткого плеча хромосомы 5. У больных при кариологическом анализе обнаруживается укорочение короткого плеча одной из хромосом группы В (рис.9)
Фенотипичекими признаками синдрома являются:
· Микроцефалия;
· Круглое «лунообразное» лицо в первые годы жизни и узкое лицо в более старшем возрасте;
· Антимонголоидный разрез глаз, эпикант, косоглазие, катаракта, очаги пигментации сетчатки, атрофия зрительных нервов;
· Плоская спинка носа, высокое небо;
· Ушные раковины деформированы;
· Синдактилия пальцев ног, косолапость, мышечная гипотония;
· Своеобразный симптом – плач при рождении, напоминающий крик кошки. Он присутствует у детей первого года жизни. Обусловлен нарушением деятельности центральной нервной системы и изменениями гортани (уменьшение надгортанника, сужение гортани, отечность слизистой оболочки).
Прогноз для жизни зависит от выраженности симптомов. Многие больные доживают до подросткового возраста. Умственная отсталость всегда глубокая. Окончательный диагноз устанавливается в результате исследования кариотипа.






