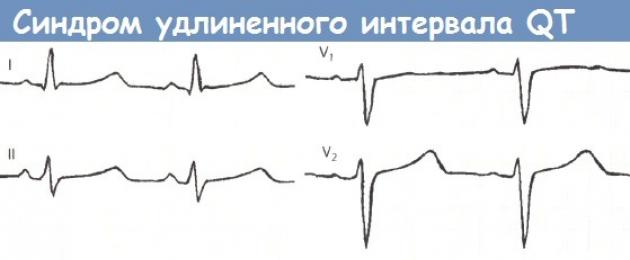
La sindrome del QT lungo è caratterizzata da 2 segni: prolungamento dell'intervallo QT (la durata dell'intervallo QT calcolato supera 0,44 s) e tachicardia ventricolare con sincope.
Oltre a queste caratteristiche, si notano un'onda U alta, un'onda T appiattita o negativa e una tachicardia sinusale.
forma congenita questa sindromeè meno frequente ed è una malattia geneticamente eterogenea, la forma acquisita è spesso dovuta alla terapia antiaritmica.
La forma congenita della sindrome del QT lungo viene trattata con bloccanti dei recettori beta-adrenergici e, in assenza di effetti terapia farmacologica se necessario, impiantare un cardioverter/defibrillatore. Con la forma acquisita è necessario innanzitutto annullare i farmaci che potrebbero causare un prolungamento dell'intervallo QT.
(sinonimo: sindrome del QT) si dividono in forma congenita, geneticamente eterogenea, e forma acquisita o indotta da farmaci. La forma congenita è estremamente rara (1 caso ogni 10.000 nati). Significato clinico La sindrome QT è che sia la sua forma congenita che quella acquisita si manifestano con tachicardia ventricolare.
I. Sindrome congenita del QT lungo (sindromi Jervell-Lange-Nielsen e Romano-Ward)
Nella patogenesi sindrome congenita del QT svolgono il ruolo di mutazioni nei geni che codificano per le proteine dei canali ionici, portando a attività insufficiente canali del potassio o maggiore attività canali del sodio. La sindrome del QT lungo può manifestarsi come sindrome di Jervell-Lange-Nielsen e sindrome di Romano-Ward.
Caratteristiche peculiari Sindrome di Jervell-Lange-Nielsen Sono:
prolungamento dell'intervallo QT
sordomutismo
episodi di svenimento e morte improvvisa.
A Sindrome di Romano-Ward non c'è sordità.
Primo manifestazioni cliniche la sindrome del QT congenita appare già in infanzia. Caratterizzato da ripetuti episodi di svenimento che compaiono sullo sfondo della simpaticotonia, ad esempio, quando il bambino piange, è stressato o urla.
Ai segni più importanti della sindrome QT relazionare:
prolungamento dell'intervallo QT, ad es. la durata dell'intervallo QT stimato supera 0,44 s (normalmente è 0,35-0,44 s)
tachicardia ventricolare (tachicardia pirouette: forma veloce e polimorfica)
bradicardia sinusale a riposo e durante esercizio
Onda T appiattita o negativa
Onda U alta o bifasica e confluenza dell'onda T e dell'onda U
dipendenza della durata dell'intervallo QT dalla frequenza cardiaca
A misurazione dell'intervallo QTè necessario prestare attenzione a non includere l'onda U (intervallo QT corretto; intervallo QTC di Bazett) nell'intervallo. L'intervallo QT relativo (ad esempio, secondo Lepeshkin o Hegglin e Holtzman) è più facile da misurare, ma il suo valore è meno accurato. Normalmente è 100±10%.
A Sindrome del QT si verifica un allungamento disomogeneo della fase di ripolarizzazione, che facilita il meccanismo di rientro dell'onda di eccitazione, contribuendo alla comparsa tachicardia ventricolare(torsione di punta, tachicardia da piroetta) e fibrillazione ventricolare.
Trattare Sindrome del QT bloccanti dei recettori beta-adrenergici e, in caso di resistenza a questi farmaci, viene impiantato un cardioverter/defibrillatore.
 Sindrome del QT lungo (sindrome di Romano-Ward).
Sindrome del QT lungo (sindrome di Romano-Ward).
FC 90 battiti al minuto, durata QT 0,42 s, durata relativa dell'intervallo QT 128%, intervallo QTC corretto allungato e pari a 0,49 s.
II. Sindrome del QT lungo acquisito
Motivi acquisiti sindrome del QT lungo, potrebbe essere diverso. Quelli di seguito sono solo quelli di maggiore importanza clinica:
farmaci antiaritmici(p. es., chinidina, sotalolo, amiodarone, aimalina, flecainide)
violazione equilibrio elettrolitico(ad esempio, ipokaliemia)
blocco del gambo PG e allargamento del complesso QRS
ipotiroidismo
cardiopatia ischemica
terapia antibiotica (p. es., eritromicina)
abuso di alcool
miocardite
emorragia cerebrale
Nei casi tipici sindrome del QT acquisito può essere associato all'uso di farmaci antiaritmici, soprattutto chinidina e sotalolo. Il significato clinico di questa sindrome è grande, dato che, come con forma congenita, la sindrome del QT acquisito è accompagnata da attacchi di tachicardia ventricolare.
Frequenza di occorrenza episodi di tachicardia ventricolare nei pazienti con sindrome del QT lungo acquisita è del 2-5%. Un tipico esempio sono la cosiddetta sincope della chinidina. I cambiamenti dell'ECG sono gli stessi di sindrome congenita Qt.
Trattamento implica innanzitutto l'abolizione del farmaco "causativo" e l'introduzione, tra l'altro, di una soluzione di lidocaina.
Caratteristiche dell'ECG nella sindrome del QT lungo:
Modifica dell'intervallo QT (intervallo QTC normale<0,44 с)
Tendenza alla tachicardia ventricolare
Forma congenita: per la sincope, in alcuni pazienti è indicato l'impianto di un cardioverter/defibrillatore
Forma acquisita: sospensione di farmaci antiaritmici (causa comune della sindrome)
La durata dell'intervallo QT riflette il tempo di ripolarizzazione dei ventricoli del cuore. La durata normale dell'intervallo QT dipende dalla frequenza cardiaca attuale. Per scopi diagnostici, il QTc assoluto più comunemente utilizzato (intervallo QT corretto), calcolato utilizzando la formula di Bazett. Questo indicatore è regolato in base alla frequenza cardiaca attuale. Esempio di calcolo QTc:
Sindrome del QT lungo (LQT, LQTS, MIM*192500)
- una malattia accompagnata da prolungamento dell'intervallo QT sull'ECG a riposo (QTc> 460 ms), sincope e alto rischio morte improvvisa a causa dello sviluppo di tachicardia ventricolare polimorfica. Le forme ereditarie di LQTS sono ereditate sia con modalità autosomica dominante che autosomica recessiva. Il prolungamento dell'intervallo QT può essere determinato geneticamente (primario) o secondario, a seguito dell'esposizione a fattori avversi(ricezione di una serie medicinali, ipokaliemia, ipomagnesemia, ipocalcemia, dieta a basso contenuto proteico e anoressia nervosa miocardite, cardiomiopatia, emorragia intracranica). La diagnosi differenziale tra forme primarie e secondarie è estremamente importante per determinare le tattiche di trattamento, valutare il rischio di aritmie potenzialmente letali e la prognosi.
 |
Recentemente, è diventato chiaro che il contributo fattori genetici in caso di prolungamento secondario dell'intervallo QT non è da sottovalutare. In una percentuale significativa di pazienti con prolungamento dell'intervallo QT indotto da farmaci, vengono rilevate le cosiddette "mutazioni silenti", o polimorfismi funzionali, negli stessi geni responsabili delle forme primarie di LQTS. I cambiamenti nella struttura dei canali ionici dei cardiomiociti in questi casi sono minimi e possono rimanere asintomatici per lungo tempo. Pertanto, una persona non può essere consapevole del fatto che alcuni farmaci ampiamente rappresentati sul mercato farmaceutico sono pericolosi per lui. Per la maggior parte delle persone, la depressione della corrente di potassio indotta dai farmaci è lieve e non causa alcuna variazione dell’ECG. Tuttavia, la combinazione di caratteristiche genetiche della struttura dei canali del potassio e dell'assunzione di farmaci può causare aritmie clinicamente significative, fino allo sviluppo di tachicardia ventricolare polimorfica "Torsade des pointes" e morte improvvisa. Pertanto, si consiglia ai pazienti che hanno registrato almeno una volta tachicardia ventricolare polimorfica causata dall'assunzione di qualsiasi farmaco di consultare un genetista. Inoltre, evitare per tutta la vita tutti i farmaci che portano al prolungamento dell’intervallo QT.
La frequenza della forma primaria della sindrome del QT lungo è di circa 1:3000. Ad oggi sono noti almeno 12 geni responsabili dello sviluppo della malattia. Una mutazione in uno qualsiasi di essi può portare allo sviluppo della malattia.
Geni responsabili dello sviluppo della sindrome del QT lungo.
| VARIANTE LQTS | GENE | POSSIBILITÀ DI DIAGNOSI DEL DNA IN RUSSIA |
| LQT1 | KCNQ1 | Tenuto |
| LQT2 | KCNH2 | Tenuto |
| LQT3 | SCN5A | Tenuto |
| LQT4 | AnkB | Non effettuato |
| LQT5 | KCNE1 | Tenuto |
| LQT6 | KCNE2 | Tenuto |
| LQT7 | KCNJ2 | Tenuto |
| LQT9 | Cav3 | Non effettuato |
| LQT10 | SCN4B | Tenuto |
| LQT11 | AKAP9 | Non effettuato |
| LQT12 | SNTA1 | Non effettuato |
È possibile richiedere una diagnosi diretta del DNA per la sindrome del QT lungo all'indirizzo centro medico Prima Medica. In base ai risultati della diagnosi del DNA viene rilasciata una conclusione scritta del genetista con l'interpretazione dei risultati. Analizzando tutti questi geni, è possibile identificare le mutazioni e stabilire la forma genetica molecolare della malattia nel 70% dei probandi. Le mutazioni in questi geni possono anche causare fibrillazione ventricolare idiopatica e sindrome della morte improvvisa del lattante (circa il 20% dei casi).
Perché è necessario effettuare la diagnostica del DNA della LQTS?
L'uso di metodi di genetica molecolare per la sindrome del QT lungo può essere cruciale nelle seguenti situazioni:
- La necessità di una conferma e/o diagnosi differenziale(ad esempio per risolvere il problema della natura primaria o secondaria dell'allungamento dell'intervallo QT).
- Identificazione delle forme asintomatiche e oligosintomatiche della malattia, ad esempio, tra i parenti di pazienti con una diagnosi consolidata. Secondo diversi autori, fino al 30% delle persone con mutazioni nei geni interessati non presentano alcun segno della malattia (compresi quelli elettrocardiografici). Allo stesso tempo, il rischio di sviluppare aritmie e morte cardiaca improvvisa rimane elevato, soprattutto se esposti a specifici fattori di rischio.
- Quando si sceglie la tattica del trattamento della malattia. È stato ora dimostrato che i pazienti con diverse forme genetiche molecolari della malattia rispondono in modo diverso al trattamento. La determinazione accurata della variante genetica molecolare della malattia consente al paziente di scegliere una terapia farmacologica adeguata, tenendo conto dell'interruzione del funzionamento di un particolare tipo di canale ionico. Efficienza vari metodi trattamento di varie varianti genetiche molecolari della sindrome LQTS. >
ICD - defibrillatore cardioverter impiantabile, PVT - tachicardia ventricolare polimorfica, ECS - pacemaker, +++ - massima efficienza dell'approccioLQT1, LQT5 LQT2, LQT6 LQT3 Sensibilità alla stimolazione simpatica +++ + - Circostanze in cui si osserva spesso la PVT paura A riposo/nel sonno Fattore specifico che provoca la sincope Nuoto Suono aspro, dopo il parto - Limitazione dell'attività fisica +++ + - b-bloccanti +++ + - Assunzione di integratori di potassio +? +++ +? Farmaci antiaritmici di classe IB (bloccanti dei canali del sodio) + ++ +++ Bloccanti dei canali del calcio ++ ++ +? Apri canali del potassio (nicorandil) + + - L'EX + + +++ ICD ++ ++ +++ - Aiuto con la pianificazione familiare. Previsioni serie malattie, un alto rischio di aritmie potenzialmente letali in assenza di una terapia adeguata, determina la rilevanza della diagnosi prenatale del DNA della LQTS. I risultati della diagnostica del DNA prenatale in famiglie con una forma genetica molecolare già consolidata di sindrome del QT lungo consentono di pianificare con successo la gestione della gravidanza, del parto e delle tattiche terapia farmacologica nel periodo postpartum.
Cosa fare se è stata identificata una mutazione?
Se tu o tuo figlio avete una mutazione che conferma la natura ereditaria della malattia, dovete ricordare quanto segue:
- È necessario discutere con un genetista i risultati di uno studio di genetica molecolare, cosa significano, quale valore clinico e prognostico possono avere.
- I tuoi parenti, anche senza Segni clinici malattie, possono essere portatori di una simile cambiamento genetico, ed essere a rischio di sviluppare aritmie pericolose per la vita. Si consiglia di discutere con loro e/o con un genetista la possibilità di consulenza e diagnosi del DNA per altri membri della vostra famiglia.
- È necessario discutere con un genetista le caratteristiche di questa variante genetica della malattia, fattori specifici rischi e come evitarli al meglio.
- Nel corso della vita è necessario evitare l'assunzione di numerosi farmaci.
- È necessaria una consultazione precoce e un follow-up a lungo termine, solitamente per tutta la vita, da parte di un aritmologo. Nel nostro Centro è attivo un programma di osservazione delle famiglie con disturbi ereditari frequenza cardiaca
La salute umana è la componente principale della normalità e vita di qualità. Ma non sempre ci sentiamo in salute. Potrebbero sorgere problemi da motivi diversi, e la loro importanza può anche essere diversa. Ad esempio, il comune raffreddore non provoca paura nelle persone, viene trattato rapidamente e non causa molti danni. salute generale. Ma se ci sono problemi con organi interni, è già più pericoloso per la vita e peggiora il nostro benessere per molto tempo.
Recentemente, molte persone lamentano problemi cardiaci e molto spesso si tratta di malattie comuni facili da trattare e diagnosticare. Ma ci sono momenti in cui un paziente ha una sindrome del QT lungo. In medicina, questo termine si riferisce a una condizione pronunciata o acquisita di una persona, accompagnata da un aumento della durata di un dato intervallo su un segmento del cardiogramma. Inoltre a questa sindrome vengono attribuiti solo allungamenti superiori a 55 ms rispetto ai valori normali. Inoltre, quando si sviluppa la malattia, gli indicatori di deviazione di questo intervallo possono essere superiori a 440 ms.
Manifestazioni
Nella maggior parte dei casi, per il paziente stesso, questa malattia è asintomatica ed è quasi impossibile rilevarla da soli. Fondamentalmente, nelle persone con questa diagnosi, i processi di ripolarizzazione e depolarizzazione vengono interrotti a causa di un cambiamento nella simmetria, cosa che può essere notata solo nel processo di ricerca, sulla base dei dati provenienti da vari tipi di apparecchiature. Il fattore principale che causa questa condizione è l’instabilità elettrica del muscolo cardiaco.
I soggetti affetti dalla sindrome del QT lungo possono sviluppare tachicardia ventricolare se il trattamento è inefficace o assente. Queste complicazioni sono molto più pericolose per la vita dei pazienti e sono dannose. condizione generale. A questo proposito, se sospetti la presenza di questa malattia, dovresti immediatamente prenderti cura della tua salute, altrimenti potrebbero verificarsi conseguenze negative. Inoltre, le complicazioni di questa malattia sono piuttosto gravi. Possono portare non solo a prestazioni ridotte e al deterioramento del benessere generale del paziente, ma anche alla morte.
Tipi
In medicina, tale deviazione è stata a lungo studiata e nel corso degli anni gli scienziati hanno potuto imparare sempre di più al riguardo. Questa malattia è divisa in due tipi, vale a dire la sindrome acquisita e quella congenita dell'intervallo QT lungo. È possibile determinare quale tipo ha un paziente solo tramite il metodo di ricerca. A disturbo congenito c'è un problema di crash codice genetico. Una volta acquisiti, vari fattori possono influenzare lo sviluppo della malattia.
Forme
Esistono anche alcuni tipi di decorso della malattia:
- forma nascosta. È caratterizzata prestazione normale intervallo durante l'esame e il primo attacco di sincope provoca una morte improvvisa.
- Si verificano attacchi di sincope, ma l'intervallo QT non risulta prolungato al momento dell'esame.
- L'allungamento dell'intervallo è isolato e non si riflette nell'anamnesi.
- La sincope si verifica con un prolungamento dell'intervallo QT superiore alla norma di 440 ms o più.
Cause
Molti fattori possono influenzare lo sviluppo di questa malattia. Quindi, ad esempio, inizia a svilupparsi a causa di malattie ereditarie, Compreso Sindrome RU. In questo caso sono molto frequenti gli attacchi di perdita di coscienza, che portano effettivamente allo sviluppo di questa malattia. Così come la sindrome E-R-L, se il paziente ha sordità congenita. Qual è la ragione di questa combinazione di sintomi e come provoca esattamente lo sviluppo della malattia, gli scienziati non sono ancora riusciti a scoprirlo.

Inoltre, la mutazione genetica può causare lo sviluppo questa malattia. Questa è la causa principale di una malattia congenita, ma in alcuni casi non appare immediatamente, ma già presente età adulta dopo le sollecitazioni. Di solito si tratta di problemi con la sintesi proteica del sodio e canali del potassio diventano fattori che provocano la sindrome del QT lungo. Il motivo potrebbe risiedere effetto collaterale prendendo alcuni farmaci. La minaccia più grande è antibiotici forti, che il paziente può assumere per curare altre malattie.

La causa della malattia può essere disordini metabolici o diete volte a ridurre le calorie negli alimenti. L'esaurimento del corpo in tali situazioni può influenzare non solo il cuore. Pertanto, è meglio coordinare tali diete con un medico ed essere costantemente sotto la sua supervisione. Lo spreco può complicare alcuni malattia cardiovascolare come la malattia ischemica o la sindrome a volte si sviluppa a causa di patologie del sistema nervoso centrale e con distonia vegetativa, così come altri disturbi del sistema nervoso autonomo.
Sintomi
Esistono segni specifici che indicano che il paziente ha la sindrome del QT lungo. I sintomi di questa malattia sono i seguenti:
- Perdita di coscienza che dura da un paio di minuti a un quarto d'ora. In alcuni casi, l'attacco può durare fino a venti minuti.
- Convulsioni in condizioni sinottiche, in apparenza simili alle crisi epilettiche, ma i processi che le provocano sono completamente diversi.
- Debolezza improvvisa nel corpo, accompagnata dall'oscuramento degli occhi.
- Forti palpitazioni anche senza attività fisica o stress emotivo.
- Dolore al petto di diversa natura, che continua durante un battito cardiaco accelerato, così come svenimenti o vertigini ad essi associati e intorpidimento delle braccia e delle gambe.
Diagnostica
Molto spesso la sindrome del QT lungo, soprattutto nei bambini, è asintomatica. In una situazione del genere, il paziente può sentirsi completamente sano e morire improvvisamente. Pertanto, se una persona è a rischio di malattia, è necessario essere regolarmente visitata da un medico per escludere la possibilità di sviluppare la malattia. Per diagnosticare una malattia, la medicina moderna utilizza diversi metodi.

Se si sospetta che un paziente abbia la sindrome del QT lungo e i problemi di salute lo indicano chiaramente, l'elettrocardiografia è lo studio più importante per determinare la malattia. Conducendolo durante un attacco, il dispositivo mostrerà segni di tachicardia ventricolare, trasformandosi in fibrillazione ventricolare. È questo metodo il principale nel determinare la forma della malattia.

C'è anche un altro studio che rivela la sindrome del QT lungo. Si effettua in giornata. Si parla quindi di monitoraggio 24 ore su 24, che consente di registrare l'attività cardiaca del paziente durante questo periodo. Al suo corpo è attaccato un piccolo apparato che registra le indicazioni del lavoro del cuore e, dopo averlo rimosso, lo specialista decifra i dati registrati dal dispositivo. Permettono di determinare se il paziente ha una bradicardia rigida grave, se cambia la morfologia dell'onda T e se ci sono disturbi nei processi di ripolarizzazione miocardica e extrasistole ventricolare.
Trattamento
Se a un paziente è stata diagnosticata la sindrome dell'intervallo QT lungo, il trattamento deve essere completo e adeguato, poiché questo è l'unico modo per prevenire lo sviluppo di complicanze pericolose per la salute e che possono essere fatali.
Terapia medica
La malattia può essere curata con farmaci antiaritmici. Un corso terapeutico adeguatamente selezionato non solo eliminerà i sintomi di questa malattia, ma stabilizzerà anche il lavoro per un lungo periodo. del sistema cardiovascolare. Questo è uno dei metodi per curare la sindrome congenita del QT lungo LQTS.
Chirurgia
Se il paziente è a rischio di aritmia pericolosa per la vita a causa di questa malattia, gli esperti raccomandano l'impianto di un pacemaker. Il suo compito è normalizzare la frequenza delle contrazioni del muscolo cardiaco. medicina moderna sviluppato dispositivi speciali che determinano deviazione patologica nell'opera del cuore. La malattia può essere causata dall'esterno. Durante lo sforzo fisico, ad esempio, il dispositivo non risponde. Ma se gli impulsi sono di natura patologica, questo normalizza il lavoro dell'organo.

L’intervento chirurgico per una malattia come la sindrome del QT lungo è semplice e abbastanza sicuro. Il pacemaker è fissato a sinistra del grande muscolo del torace. Da esso escono gli elettrodi, che i chirurghi fissano nell'area richiesta, facendoli passare attraverso vena succlavia. Il dispositivo può essere configurato tramite il programmatore. Con esso è possibile modificare i parametri della stimolazione cardiaca, a seconda delle caratteristiche personali del paziente. Il dispositivo si accenderà ogni volta che il lavoro del muscolo cardiaco supera i parametri specificati.
Conclusione
Questa malattia non è sempre possibile diagnosticare, poiché raramente si manifesta in modo pronunciato. Ma allo stesso tempo la minaccia per la salute del paziente è molto grande. Pertanto, se esiste almeno un leggero rischio che si verifichi, vale la pena sottoporsi costantemente a esami e consultare specialisti.

Se la diagnosi è confermata, è necessario un trattamento completo e completo di questa malattia, perché può essere fatale.
L'articolo è dedicato alla sindrome ECG congenita e acquisita intervallo prolungato QT, così come Amiodarone, come i più comuni motivo medico questo stato.
La sindrome del QT lungo è una combinazione di un intervallo QT lungo su un ECG standard e in pericolo di vita tachicardie ventricolari polimorfiche (torsade de pointes - "pirouette"). I parossismi di tachicardia ventricolare del tipo "piroetta" si manifestano clinicamente con episodi di perdita di coscienza e spesso terminano con la fibrillazione ventricolare, che è la causa diretta della morte improvvisa.
La durata dell'intervallo QT dipende dalla frequenza cardiaca e dal sesso del paziente. Pertanto, non utilizzano il valore assoluto, ma il valore corretto dell'intervallo QT (QTc), che viene calcolato utilizzando la formula di Bazett:
dove: RR è la distanza tra onde R adiacenti sull'ECG in sec. ;
K = 0,37 per gli uomini e K = 0,40 per le donne.
Il prolungamento dell'intervallo QT viene diagnosticato se la durata del QTc supera 0,44 s.
È stato stabilito che sia le forme congenite che quelle acquisite di prolungamento dell'intervallo QT sono predittori di aritmie fatali, che, a loro volta, portano alla morte improvvisa dei pazienti.
IN l'anno scorso Molta attenzione è rivolta allo studio della variabilità (dispersione) dell'intervallo QT - un indicatore della disomogeneità dei processi di ripolarizzazione, poiché una maggiore dispersione dell'intervallo QT è anche un predittore dello sviluppo di una serie di gravi violazioni ritmo, compresa la morte improvvisa. La varianza dell'intervallo QT è la differenza tra i valori massimo e minimo dell'intervallo QT, misurata in 12 standard Derivazioni dell'ECG: D QT = QTmax-QTmin.
Pertanto, non c’è consenso sul limite superiore valori normali variazione dell’intervallo QT corretto. Secondo alcuni autori, un QTcd superiore a 45 è un predittore di tachiaritmia ventricolare, altri ricercatori suggeriscono che il limite superiore del QTcd normale sia di 70 ms e addirittura 125 ms.
Ce ne sono due più studiati meccanismo patogenetico Aritmie nella sindrome del QT lungo. Il primo è il meccanismo dei "disturbi intracardiaci" della ripolarizzazione miocardica, vale a dire ipersensibilità miocardio all’effetto aritmogeno delle catecolamine. Secondo meccanismo fisiopatologico: lo squilibrio innervazione simpatica(diminuzione dell'innervazione simpatica destra dovuta a debolezza o sottosviluppo del ganglio stellato destro). Questo concetto è supportato da modelli animali (prolungamento dell'intervallo QT dopo stellectomia del lato destro) e dai risultati della stellectomia del lato sinistro nel trattamento. forme resistenti prolungamento dell'intervallo QT.
La frequenza di rilevamento del prolungamento dell'intervallo QT negli individui con prolasso delle valvole mitrale e/o tricuspide raggiunge il 33%. Secondo la maggior parte dei ricercatori, il prolasso della valvola mitrale è una delle manifestazioni della displasia congenita. tessuto connettivo. Tra le altre manifestazioni di "debolezza del tessuto connettivo" - aumento dell'estensibilità della pelle, tipo di corpo astenico, deformità a forma di imbuto Petto, scoliosi, piedi piatti, sindrome da ipermobilità articolare, miopia, vene varicose vene, ernie. Numerosi ricercatori hanno identificato una relazione tra l’aumento della variabilità dell’intervallo QT e la profondità del prolasso e/o la presenza di cambiamenti strutturali(degenerazione mixomatosa) dei lembi della valvola mitrale. Uno dei motivi principali per la formazione del prolungamento dell'intervallo QT nei soggetti con prolasso della valvola mitrale è una carenza di magnesio geneticamente predeterminata o acquisita.
Il prolungamento acquisito dell'intervallo QT può verificarsi nella cardiosclerosi aterosclerotica o post-infartuale, nella cardiomiopatia, contro e dopo mio- o pericardite. Un aumento della dispersione dell'intervallo QT (più di 47 ms) può anche essere un predittore dello sviluppo di sincope aritmogena nei pazienti con difetti aortici cuori.
Si può osservare anche un prolungamento dell'intervallo QT bradicardia sinusale, blocco atrioventricolare, insufficienza cerebrovascolare cronica e tumori cerebrali. Casi acuti il prolungamento dell'intervallo QT può verificarsi anche in caso di lesioni (toraciche, craniocerebrali).
La neuropatia autonomica aumenta anche l'intervallo QT e la sua dispersione, quindi queste sindromi si verificano nei pazienti diabete I e II tipo.
Il prolungamento dell'intervallo QT può verificarsi con squilibrio elettrolitico con ipokaliemia, ipocalcemia, ipomagnesiemia. Tali condizioni sorgono sotto l'influenza di molte ragioni, ad esempio quando uso a lungo termine diuretici, in particolare i diuretici dell'ansa (furosemide). Viene descritto lo sviluppo di tachicardia ventricolare del tipo "piroetta" sullo sfondo del prolungamento dell'intervallo QT con esito fatale nelle donne che seguivano una dieta ipoproteica per ridurre il peso corporeo.
Il prolungamento del QT è ben noto ischemia acuta miocardio e infarto miocardico. L'aumento persistente (più di 5 giorni) dell'intervallo QT, soprattutto se combinato con extrasistoli ventricolari precoci, ha un prognostico sfavorevole. Questi pazienti hanno mostrato un aumento significativo (5-6 volte) del rischio di morte improvvisa.
Nella patogenesi del prolungamento dell’intervallo QT nell’infarto miocardico acuto gioca sicuramente un ruolo l’iperisimpaticotonia, questo spiegano molti autori alta efficienza b-bloccanti in questi pazienti. Inoltre, lo sviluppo di questa sindrome si basa su disturbi elettrolitici in particolare la carenza di magnesio. I risultati di molti studi indicano che fino al 90% dei pazienti con infarto acuto il miocardio è carente di magnesio. È stata riscontrata anche una correlazione inversa tra il livello di magnesio nel sangue (siero ed eritrociti) e l'intervallo QT e la sua dispersione nei pazienti con infarto miocardico acuto.
Nei pazienti con prolasso della valvola mitrale idiopatico, il trattamento deve iniziare con l'uso di preparati orali di magnesio (Magnerot 2 compresse 3 volte al giorno per almeno 6 mesi), poiché la carenza tissutale di magnesio è considerata uno dei principali meccanismi fisiopatologici per la formazione di entrambi. la sindrome di allungamento dell'intervallo QT e "debolezza" del tessuto connettivo. In questi individui, dopo il trattamento con preparati a base di magnesio, non solo l'intervallo QT si normalizza, ma diminuisce anche la profondità del prolasso delle cuspidi della valvola mitrale, la frequenza extrasistoli ventricolari, gravità delle manifestazioni cliniche (sindrome distonia vegetativa, sintomi emorragici e così via.). Se il trattamento farmaci per via orale niente magnesio dopo 6 mesi pieno effetto viene mostrata l'aggiunta di b-bloccanti.
Un'altra causa importante del prolungamento dell'intervallo QT è l'uso di farmaci speciali, uno di questi farmaci più comunemente usati in pratica clinicaè Amiodarone (Cordarone).
L'amiodarone appartiene ai farmaci antiaritmici di classe III (una classe di inibitori della ripolarizzazione) e possiede un meccanismo d'azione antiaritmico unico, poiché oltre alle proprietà degli antiaritmici di classe III (blocco dei canali del potassio), ha gli effetti dei farmaci antiaritmici di classe I (blocco dei canali del potassio). blocco dei canali del calcio), farmaci antiaritmici di classe IV (blocco dei canali del calcio). ) e azione beta-bloccante non competitiva.
Oltre all'azione antiaritmica, ha effetti antianginosi, dilatatori delle coronarie, alfa e beta adrenobloccanti.
Proprietà antiaritmiche:
- un aumento della durata della 3a fase del potenziale d'azione dei cardiomiociti, dovuto principalmente al blocco della corrente ionica nei canali del potassio (l'effetto di un antiaritmico di classe III secondo la classificazione di Williams);
- diminuzione dell'automatismo nodo del seno portando ad una diminuzione della frequenza cardiaca;
- blocco non competitivo dei recettori alfa e beta adrenergici;
Descrizione
- decelerazione della conduzione senoatriale, atriale e atrioventricolare, più pronunciata con la tachicardia;
- nessun cambiamento nella conduzione ventricolare;
- un aumento dei periodi refrattari e una diminuzione dell'eccitabilità del miocardio degli atri e dei ventricoli, nonché un aumento del periodo refrattario del nodo atrioventricolare;
- rallentamento della conduzione e aumento della durata del periodo refrattario in ulteriori fasci di conduzione atrioventricolare.
Altri effetti:
- nessun effetto inotropo negativo se assunto per via orale;
- diminuzione del consumo di ossigeno da parte del miocardio a causa di una moderata diminuzione resistenza periferica e frequenza cardiaca;
- un aumento del flusso sanguigno coronarico dovuto ad un effetto diretto sulla muscolatura liscia delle arterie coronarie;
- mantenimento gittata cardiaca riducendo la pressione nell'aorta e riducendo la resistenza periferica;
- influenza sul metabolismo degli ormoni tiroidei: inibizione della conversione di T3 in T4 (blocco della tiroxina-5-deiodinasi) e blocco della cattura di questi ormoni da parte di cardiociti ed epatociti, con conseguente indebolimento dell'effetto stimolante degli ormoni tiroidei sull'organismo miocardio.
Gli effetti terapeutici si osservano in media una settimana dopo l'inizio del farmaco (da diversi giorni a due settimane). Dopo aver interrotto la sua assunzione, l'amiodarone viene determinato nel plasma sanguigno per 9 mesi. Dovrebbe essere presa in considerazione la possibilità di mantenere l'azione farmacodinamica dell'amiodarone per 10-30 giorni dopo la sua sospensione.
Ciascuna dose di amiodarone (200 mg) contiene 75 mg di iodio.
Indicazioni per l'uso
Prevenzione delle ricadute
- Aritmie ventricolari pericolose per la vita, comprese tachicardia ventricolare e fibrillazione ventricolare (il trattamento deve essere iniziato in un ospedale con stretto monitoraggio cardiaco).
- Tachicardia parossistica sopraventricolare:
- attacchi documentati di ricorrenti sopraventricolari sostenuti tachicardia parossistica in pazienti con cardiopatia organica;
- attacchi documentati di tachicardia parossistica sopraventricolare sostenuta ricorrente in pazienti senza malattie organiche cuore, quando i farmaci antiaritmici di altre classi non sono efficaci o vi sono controindicazioni al loro uso;
- attacchi documentati di tachicardia parossistica sopraventricolare sostenuta ricorrente in pazienti con sindrome di Wolff-Parkinson-White.
- Fibrillazione atriale (fibrillazione atriale) e flutter atriale
Prevenzione della morte aritmica improvvisa nei pazienti ad alto rischio
- Pazienti dopo di recente infarto miocardico miocardio con più di 10 extrasistoli ventricolari all'ora, manifestazioni cliniche di insufficienza cardiaca cronica e una ridotta frazione di eiezione ventricolare sinistra (meno del 40%).
L'amiodarone può essere utilizzato nel trattamento delle aritmie nei pazienti con cardiopatia ischemica e/o disfunzione ventricolare sinistra
Per i pazienti con insufficienza cardiaca cronica, l’amiodarone è l’unico farmaco antiaritmico approvato. Ciò è dovuto al fatto che altri farmaci in questa categoria di pazienti aumentano il rischio di morte cardiaca improvvisa o deprimono l'emodinamica.
In presenza di malattia coronarica cuore, il farmaco d’elezione è il sotalolo, che è noto essere un bloccante β-adrenergico di 1/3. Ma con la sua inefficienza, ancora una volta abbiamo a nostra disposizione solo l’amiodarone. Per quanto riguarda i pazienti con ipertensione arteriosa, quindi dal loro numero, a loro volta, si distinguono i pazienti con ipertrofia grave e inespressa del ventricolo sinistro. Se l'ipertrofia è piccola (nelle Linee Guida del 2001 lo spessore della parete del ventricolo sinistro è inferiore a 14 mm), il farmaco d'elezione è il propafenone, ma se è inefficace, come sempre, l'amiodarone (insieme al sotalolo). Infine, nell’ipertrofia ventricolare sinistra grave, come nell’insufficienza cardiaca cronica, l’amiodarone è l’unico farmaco possibile.
20 luglio 2018 Nessun commento
La sindrome del QT lungo è una malattia congenita caratterizzata dal prolungamento dell'intervallo QT sull'elettrocardiogramma (ECG) e da una tendenza alla tachicardia ventricolare, che può portare a sincope, arresto cardiaco o morte cardiaca improvvisa (SCD). Vedi l'immagine qui sotto.
L'intervallo QT sull'ECG, misurato dall'inizio del complesso QRS alla fine dell'onda T, rappresenta la durata dell'attivazione e del recupero del miocardio ventricolare. Un intervallo QT corretto per la frequenza cardiaca superiore a 0,44 secondi è generalmente considerato anormale, sebbene un QTc normale possa essere più lungo nelle donne (fino a 0,46 secondi). La formula di Bazett è la formula più comunemente utilizzata per calcolare il QTc, nel seguente modo: QTc = QT / Radice quadrata Intervallo R-R (in secondi).
Per misurare con precisione l'intervallo QT, la relazione tra QT e intervallo R-R deve essere riproducibile. Questo problema è particolarmente importante quando la frequenza cardiaca è inferiore a 50 battiti al minuto (bpm) o superiore a 120 bpm e quando atleti o bambini hanno segnato Variabilità R-R. In questi casi, a lungo Registrazioni dell'ECG e molteplici dimensioni. L'intervallo QT più lungo si osserva solitamente nelle derivazioni atriali destre. Quando il cambiamento marcato è presente in intervallo R-R(fibrillazione atriale, ectopia), la correzione dell'intervallo QT è difficile da determinare con precisione.

segni e sintomi
La sindrome del QT lungo viene solitamente diagnosticata dopo che una persona ha uno svenimento o attacco di cuore. In alcune situazioni, questa condizione viene diagnosticata dopo la morte improvvisa di un membro della famiglia. In alcune persone, la diagnosi viene posta quando l'ECG mostra un prolungamento dell'intervallo QT.
Diagnostica
I risultati dell'esame obiettivo solitamente non indicano una diagnosi di sindrome del QT lungo, ma alcune persone possono avere una bradicardia eccessiva per la loro età e alcuni pazienti possono avere una perdita dell'udito (sordità congenita), suggerendo la possibilità della sindrome di Jervell e Lange-Nielsen. Anomalie scheletriche come bassa statura e scoliosi sono presenti nella sindrome di Andersen. difetti di nascita cuori, cognitivi e problemi comportamentali, nella sindrome di Timothy si possono osservare malattie dell'apparato muscolo-scheletrico e disfunzioni immunitarie.
Ricerca
I test diagnostici nelle persone con sospetta sindrome includono quanto segue:
- Misurazione del livello di potassio e magnesio nel siero;
- Studio della funzione tiroidea;
- Test provocativi farmacologici con epinefrina o isoproterenolo;
- Elettrocardiografia del paziente e dei familiari;
- Test genetici del paziente e dei familiari.
Un intervallo QT corretto prolungato in risposta ad un test in piedi, che è associato ad un aumento del tono simpatico, può fornire maggiori informazioni diagnostiche nei pazienti con la sindrome. Questo aumento del QT dovuto alla posizione eretta può persistere anche dopo che la frequenza cardiaca è tornata alla normalità.
Trattamento
Nessun trattamento può eliminare la causa della sindrome del QT lungo. Le misure terapeutiche antiadrenergiche (p. es., l'uso di beta-bloccanti, stellectomia cerucotracale sinistra) e la terapia con dispositivi (p. es., l'uso di pacemaker, defibrillatori cardioverter impiantabili) mirano a ridurre il rischio e la mortalità di attacchi cardiaci.
Medico
Gli agenti beta-bloccanti adrenergici sono farmaci che possono essere prescritti per trattare la sindrome e comprendono i seguenti farmaci:
- Nadolol
- propranololo
- metoprololo
- Atenololo
Detto questo, Nadolol è il beta-bloccante preferito, che deve essere utilizzato alla dose di 1-1,5 mg/kg/die (una volta al giorno per i pazienti di età superiore a 12 anni, due volte al giorno per i più giovani).
Chirurgia
La chirurgia per le persone con sindrome del QT lungo può includere le seguenti procedure:
Impianto di defibrillatori cardioverter
Posizionamento del pacemaker
Stellectomia cervicotoracica sinistra
Le persone che hanno la sindrome dovrebbero evitare di partecipare a competizioni sportive e di esibirsi in modo pesante esercizio fisico e cerca di non evitare lo stress emotivo.
Inoltre, dovrebbero essere evitati anche i seguenti farmaci:
Anestetici o farmaci per l’asma (come l’adrenalina)
Antistaminici (p. es., difenidramina, terfenadina e astemizolo)
Antibiotici (p. es., eritromicina, trimetoprim e sulfametossazolo, pentamidina)
Farmaci cardiaci (p. es., chinidina, procainamide, disopiramide, sotalolo, probucolo, bepridil, dofetilide, ibutilide)
Farmaci gastrointestinali (p. es., cisapride)
Antifungini (p. es., ketoconazolo, fluconazolo, itraconazolo)
Farmaci psicotropi (p. es., antidepressivi triciclici, derivati della fenotiazina, butirrofenoni, benzisossazolo, difenilbutilpiperidina)
Farmaci che perdono potassio (p. es., indapamide, altri diuretici, farmaci per il vomito/diarrea)
Cause
L'intervallo QT rappresenta la durata dell'attivazione e del recupero del miocardio ventricolare. Il recupero prolungato dalla stimolazione elettrica aumenta la probabilità di refrattarietà dispersiva, dove parti del miocardio possono essere immuni alla successiva depolarizzazione.
CON punto fisiologico della vista, la dispersione avviene durante la ripolarizzazione tra i tre strati del cuore e la fase di ripolarizzazione tende ad aumentare nel miocardio medio. Questo è il motivo per cui l'onda T è solitamente ampia e l'intervallo Tpeak-Tend (Tp-e) rappresenta la dispersione transmurale della ripolarizzazione. A sindrome prolungata Il QT aumenta e crea la funzionalità per la riattivazione transmurale.
L'ipokaliemia, l'ipocalcemia e l'uso di diuretici dell'ansa sono fattori di rischio per il prolungamento dell'intervallo QT.
La sindrome è divisa in due varianti cliniche: sindrome di Romano-Ward (origine familiare con trasmissione autosomica dominante, prolungamento dell'intervallo QT e tachicardie ventricolari) o sindrome di Jervell e Lang-Nielsen (origine familiare con trasmissione autosomica recessiva, sordità congenita, prolungamento dell'intervallo QT e aritmie ventricolari). Sono state descritte altre due sindromi: la sindrome di Andersen e la sindrome di Timothy, anche se tra gli scienziati si discute se queste debbano essere incluse nella sindrome del QT lungo.
Tachiaritmia
Il prolungamento dell'intervallo QT può portare a tachicardia ventricolare polimorfica, che a sua volta può portare a fibrillazione ventricolare e morte cardiaca improvvisa. È opinione diffusa che la torsione di punta sia attivata dalla riattivazione dei canali del calcio, dalla riattivazione della corrente di sodio ritardata o da una diminuzione della corrente della camera, che porta a una post-depolarizzazione precoce, in uno stato di aumentata dispersione transmurale della ripolarizzazione, solitamente associato a un intervallo QT prolungato, funge da substrato ausiliario funzionale per mantenere la tachicardia.
La dispersione transmurale della ripolarizzazione non solo fornisce un substrato per il meccanismo di rientro, ma aumenta anche la probabilità di una post-depolarizzazione precoce, l’evento scatenante della tachiaritmia, prolungando la finestra temporale in cui i canali del calcio rimangono aperti. Qualunque condizione aggiuntiva, che accelera la riattivazione dei canali del calcio (ad esempio, aumento del tono simpatico), aumenta il rischio di post-depolarizzazione precoce.
Genetica
È noto che la sindrome del QT lungo è causata da mutazioni nei geni dei canali cardiaci del potassio, del sodio o del calcio; sono stati identificati almeno 10 geni. Sulla base di questo background genetico, sono caratterizzati 6 tipi di sindrome di Romano-Ward, sindrome di Andersen di tipo 1 e sindrome di Timothy di tipo 1 e 2 tipi di sindrome di Jervell-Lange-Nielsen.
La sindrome è il risultato di mutazioni nei geni che codificano per le proteine dei canali ionici cardiaci che causano una cinetica anomala dei canali ionici. L'apertura ridotta del canale del potassio nella sindrome di Jervell-Lange-Nielsen di tipo 1, tipo 2, tipo 5, tipo 6, tipo 1 e tipo 1 e la chiusura ritardata del canale del sodio nella sindrome di tipo 3 ricaricano la cellula miocardica con ioni positivi .
Nelle persone affette dalla sindrome, una varietà di stimoli adrenergici, tra cui esercizio fisico, emozioni, forte rumore e il nuoto, possono accelerare l'aritmia feedback. Tuttavia, le aritmie possono verificarsi anche senza tali condizioni preliminari.
Prolungamento dell’intervallo QT indotto da farmaci
Il prolungamento secondario (indotto da farmaci) dell’intervallo QT può anche aumentare il rischio di tachiaritmie ventricolari e di morte cardiaca improvvisa. Il meccanismo ionico è simile al meccanismo ionico osservato nella sindrome congenita (cioè blocco interno del rilascio di potassio).
Oltre ai farmaci che possono potenzialmente prolungare l’intervallo QT, molti altri fattori giocano un ruolo in questo disturbo. Fattori importanti I rischi per il prolungamento dell’intervallo QT indotto da farmaci sono i seguenti:
Disturbi elettrolitici (ipokaliemia e ipomagnesiemia)
Ipotermia
Funzione tiroidea anormale
Cardiopatie strutturali
Bradicardia
Il prolungamento dell'intervallo QT da parte di farmaci può anche avere un background genetico costituito da una predisposizione dei canali ionici a una cinetica anormale causata da una mutazione genetica o da un polimorfismo. Tuttavia, non ci sono dati sufficienti per affermare che in tutti i pazienti con prolungamento dell'intervallo QT causato da medicinale, esiste una condizionalità genetica della sindrome.
Previsione
La prognosi è buona per le persone affette dalla sindrome, che vengono trattate con beta-bloccanti (e, se necessario, con altri misure terapeutiche). Fortunatamente, gli episodi di torsione di punta sono solitamente autolimitanti nei pazienti con sindrome del QT; solo il 4-5% circa degli attacchi cardiaci sono fatali.
Le persone ad alto rischio (cioè coloro che hanno avuto un arresto cardiaco o attacchi cardiaci ricorrenti nonostante la terapia con beta-bloccanti) hanno un rischio significativamente maggiore di morte cardiaca improvvisa. Per il trattamento di tali pazienti viene utilizzato un defibrillatore cardioverter impiantabile; la prognosi dopo l'impianto di ICD è buona.
Mortalità, morbilità e risposta a trattamento farmacologico differire in tipi diversi sindrome.
La sindrome del QT lungo può portare a sincope, morte cardiaca improvvisa, che di solito si verifica nei giovani sani.
Sebbene la morte cardiaca improvvisa si verifichi solitamente nei pazienti sintomatici, può verificarsi anche durante il primo episodio di sincope in circa il 30% dei pazienti. Ciò sottolinea l’importanza di diagnosticare la sindrome nel periodo presintomatico. A seconda del tipo di mutazione presente, può verificarsi morte cardiaca improvvisa attività fisica, stress emotivo, riposare o dormire. La sindrome di tipo 4 è associata a fibrillazione parossistica atri.
Studi scientifici hanno dimostrato una migliore risposta al trattamento farmacologico con una ridotta incidenza di morte cardiaca improvvisa nella sindrome QT di tipo 1 e 2 rispetto a quella di tipo 3.
I deficit neurologici dopo un arresto cardiaco interrotto possono complicare il decorso clinico dei pazienti dopo una rianimazione riuscita.
Video: sindrome del QT lungo
- In contatto con 0
- Google Plus 0
- OK 0
- Facebook 0






